Hi Sander, that’s all compelling; I’ll just leave it at my continued reluctance to conclude that the process of looking for covariate imbalance is objective and well-defined.
1 Like
A sincere thank-you to all participating in this thread. Having said that…I rest my case.
My layperson’s gut instinct is to agree (as does, apparently, FDA) with Drs.Harrell and Senn on this question, for exactly the reason laid out in the above quote. But we also have a prominent textbook of epidemiology, written by top experts in that field, arguing the opposite stance, using math and symbols that are inscrutable to 99.9% of the population.
All this is quite problematic for the medical field, which depends on consensus around RCT interpretation to recommend (or not) certain treatments for patients. Whenever a stakeholder wants to argue for or against post-hoc covariate adjustment (which can determine whether a trial result ends up being “statistically significant” or “non-significant”), they will be able to find an expert who supports them. Hence this post. Of course, it feels like the question would be moot if the concept of “statistical significance” were abandoned, but, realistically, this isn’t going to happen any time soon (as this is the bar regulators use to decide which therapies to approve).
3 Likes
This is indeed an important point to further popularize in medicine: statistics is essentially applied epistemology (or computational epistemology). Ask five statisticians for their opinion, and we’ll get six different answers. And that is OK. Senn and Hernan do agree on key fundamentals. Whenever they disagree we can explore both approaches and see if their results are meaningfully different, which assumptions are more plausible, and what trade-offs do we prioritize depending on the problem at hand. This is also in line with clinical practice where multiple answers may be available for the same scenario ![]()
2 Likes
I’m all for a conciliatory approach in situations where there is no “ground truth.” And I completely agree that clinical practice is rife with such situations.
There are more grey areas in medicine than sunlit ones. As a result, two specialists might make different recommendations to the same patient and the patient then needs to weigh pros/cons of each option before making a decision. But it’s absolutely essential, in these situations, that each specialist be able to acknowledge that the other might have a differing view, and that reasonable arguments can be made for either approach. I recall a troubling case, several years ago, when, in a clinical scenario with two options for the patient, neither good, one specialist effectively declared that the other (in the same field) had been incompetent (though this was most definitely not the case). This was incredibly distressing to the family, who felt that they had been provided with the wrong advice.
The above clinical example illustrates why, in situations where expert opinion, rather than ground truth, underlies a recommendation, it’s essential that both sides of an argument be presented, transparently. What we don’t need, is for one side to persist in saying that the other is “incorrect,” when the argument is, in fact, founded in opinion, rather than ground truth. This absolutism is what the epi/stats literature seems to devolve into, more often than it probably should- and this is the attitude that is so corrosive for students (the “patients” in this scenario).
Put another way, experts in these two fields would ideally agree, at the very least, going forward, to distinguish “poh-tay-toe”/“poh-tah-toe” types of disagreements from creationism/evolution types of disagreements.
4 Likes
Well said! Important and feasible to do so.
1 Like
Hi Frank. Just to be clear, I was not discussing “looking for covariate imbalance” so I’m not sure why you mentioned it in your reply. Let’s see if we actually disagree about anything in this topic:
Data analysis is always subjective insofar as the goals of the analysis will vary across analysts (often without them realizing their divergence). DeFinetti and Neyman agreed on this point, as does anyone who understands what math does not do (e.g., it does not pick loss functions). Thus I think “objective” is at best a synonym for a method precise enough to replicate exactly with a program; that doesn’t mean it is optimal or even sensible.
I could easily make “looking for imbalance” mathematically precise and well-defined enough to program. But formalizing “looking for imbalance” would be pointless, as I think we (including me, Senn, you, and likely Hernan too) would agree that covariate imbalance is not a goal of data analysis, at least if no fraud is suspected. Sensible formal goals would include optimizing estimation in some fashion, as in uniformly most accurate and minimum-loss estimation of effects. For those, the process of specifying loss will precisely define the effect measure (such as a causal risk difference in the randomized population).
By combining precise estimation goals with precise specification of the behavior of the data generator (which randomization helps provide), one can produce precise and well-defined distributions for effects, such as compatibility distributions (P-value functions) and posterior distributions. Data-optimized methods will use available covariate information - albeit in ways unfamiliar in RCTs, such as TML estimation. The methods are not just “looking at imbalance”; rather they are looking at the entire data distribution (treatments, covariates, outcomes) in light of the generator information (especially causal assumptions such as treatment randomization, which makes treatment exogenous).
Regression textbooks (including yours) describe some very sophisticated prediction methods that could be applied to improve effect estimation from RCTs. This can be done simply by using the methods with baseline covariates to predict potential outcomes (as in g-computation). Please explain if and why you would hesitate to use those methods to exploit baseline covariate information in an RCT, when in your textbook you don’t seem to hesitate to use the methods for clinical prediction from the same sort of baseline covariates.
2 Likes
That’s the only part that I halfway disagree with Sander. Since the list of covariates collected is somewhat arbitrary, and the sublist of those collected that experts would care to adjust outcome heterogeneity for is also somewhat arbitrary, fully programming this may be a bit difficult.
1 Like
Sorry, I’m still not sure what you are disagreeing with.
My discussion was predicated on using available covariates, which for practical purposes means those measured and then coded in the analysis data base. I think that restriction is implicit in just about every stat method in textbooks including yours (the exceptions are uncommon and mostly involve modeling unmeasured covariates in sensitivity and bias analyses). A general conclusion from all this is that, even if an approach (like “looking for imbalance”) is rejectable on reasonable grounds, we should not criticize it for problems that are intrinsic to the application and thus must be dealt with by all approaches.
When you build a clinical-prediction model you face exactly the same “arbitrariness” about which covariates to use as do those looking for imbalance or those looking to adjust effect estimates (whether in an RCT or a nonexperimental comparison). All these tasks require external (“prior”) input about which available covariates to put in the candidate list, information that is usually expressed verbally but can be formalized.
This list-specification problem is unavoidable; in particular it is not avoided by declaring no adjustment is necessary (or worse, no adjustment should be done) in RCTs: That approach is simply the extreme of ignoring all baseline covariate information and thus defaulting to an empty candidate list; it thus corresponds to clinical-prediction modeling in which only the intercept is allowed in the model.
At the other extreme, I have seen statisticians recommend propensity scores containing all covariates no matter how long the candidate list, and then use the scores for adjustment in some conventional way (e.g., stratification, matching, or inverse-probability weighting). I have also seen stepwise propensity modeling, which is an automation of “looking for imbalance” that can be implemented quickly with ordinary software. Both approaches are simply awful in terms of ordinary optimization criteria, e.g., both can blow up the variances of effect estimators, and stepwise selection can leave enormous confounding in observational estimates. But they are no more vague, arbitrary, or ill-defined than any programmable method for clinical prediction modeling or covariate adjustment.
The general problems all methods face is that of determining what information to use in the analysis and how to use it. It is a hard problem, but formal tools for dealing with it are available. For example, in causal analyses the qualitative part of the information can be encoded in the presence and direction of open paths in a causal diagram, which provides a minimal screening for candidacy (e.g., it would exclude the patient ID number but include their age). The quantitative external information (if any) can be encoded in treatment and outcome models with penalty functions or prior distributions on their parameters. Those models should be checked to ensure they are consistent with the diagram; examples of inconsistencies include models that ignore collider bias or the Table-2 (mediation) fallacy. Major portions of the Hernan & Robins book are devoted to explaining such inconsistencies in common applications of statistics, and then providing methods for building consistent causal models from the candidate covariate list.
Any disagreement now?
4 Likes
Dr.Greenland
I apologize, in advance, for not being able to discuss this topic using more technical language- please bear with me here. Some of your talks betray a profound frustration with the state of scientific research in general and research criticism in particular. You note that the result of any given study is likely to be a LOT more uncertain than most people realize. Since statistical techniques are often misapplied and their underlying assumptions/limitations poorly understood, our inferences will often end up being incorrect. “Dichotomania,” “nullism,” and poor use of language (e.g., “confidence” interval rather than “compatibility” interval) are a few examples of widespread problems. Cognitive/intellectual biases are universal, yet inadequately acknowledged and motivated reasoning often seems to underpin study interpretation. I hope this paragraph hasn’t misrepresented your views.
While a lot of these errors can affect both observational studies and RCTs, their consequences will differ in each setting. And I wonder if it is statisticians’ experience working within more highly regulated settings (e.g., designing/analyzing RCTs for pharmaceutical companies or drug regulators) that drives their preference for pre-specification of analytic choices, with the goal of minimizing the opportunity for cognitive biases to impact results.
Let’s consider the consequences, for patients, of “getting it wrong,” both in the observational setting and the experimental setting.
As you know, in medicine, observational evidence is often used to identify less common treatment-related harms (harms that might be too rare or delayed to manifest with interpretable frequency in the RCTs used for drug approval). In this context, the “nullism” error (i.e., suggesting that a drug is “safe” just because the compatibility interval around a harm signal crosses the null) can lead to the discounting of a safety signal. The result is potential failure to prevent harm to a lot of people. The converse of nullism in this setting is “crying wolf” (i.e., flagging a potentially serious treatment-related harm when that harm doesn’t actually exist). The consequence of crying wolf is unnecessary patient anxiety and potentially widespread discontinuation of therapies that were providing important benefits. Drug companies like nullism when it’s applied to observational drug safety studies- they don’t like bad press for their products. But it’s equally important to acknowledge that at least some drug safety researchers like crying wolf. Administrative databases are a goldmine of potentially headline-grabbing associations. The practice of dredging a database to identify frightening safety signals, followed by post-hoc generation of a biological rationale to explain why the observed associations must be causal in nature, is a great way to keep a career humming along. An observational drug safety researcher whose incentives are misaligned (either consciously or subconsciously e.g., after a previous traumatic run-in with a defensive pharmaceutical company) will not find it hard to make a good therapy look bad using these methods if career advancement (or revenge) is his/her main motivation.
As everyone knows, RCTs are the primary method used in medicine to identify treatment-related efficacy. In the experimental context, nullism (i.e., concluding that efficacy has not been demonstrated simply because the compatibility interval around the point estimate crosses the null, even if that interval does not exclude a clinically important benefit) can lead to premature discarding of a therapy that might otherwise help a lot of people. The converse of nullism, in an RCT context, is falsely concluding that intrinsic therapeutic efficacy is present when it’s not; this scenario is very bad since it can lead to approval of dud drugs (“regulator’s regret”). If such drugs slip under the regulatory wire, patients will incur no benefit- only the risk of side effects. Money (often massive sums, if the drug is expensive and/or widely used) and time will be wasted and more efficacious therapies will be forsaken. Drug companies do everything possible to avoid nullism in the RCT context (including, at times, spinning unimpressive trial results unscrupulously to make them seem positive). They don’t like bad press for their drugs in development, since they have already spent so much money on them. But while trialists, like all researchers, will have intellectual biases, it’s hard to make a dud treatment look stellar enough for approval through nefarious means without at least a few subject matter/methodologic experts and ethics boards and regulators noticing. In general, bad actors will have more people looking over their shoulder in the RCT context; sunlight is the best disinfectant.
In summary, biases of various types are widespread in medical research. If we expect observational researchers to pre-specify their DAGs in order to justify their subsequent analytic choices, why would it be unreasonable to require that trialists prespecify which covariates they will adjust for in the analysis phase of a trial- especially if those analytic choices can potentially mean the difference between statistical significance and insignificance (and therefore drug approval versus rejection and massive profit versus massive financial loss)? Both of these recommendations, at their roots, aim to limit subsequent analytical flexibility so as to minimize the impact of cognitive biases and misaligned incentives on decisions that affect patients’ lives.
2 Likes
Are there ballpark minimum sample sizes for RCTs testing interventions in human diseases that can to be taken seriously? I suppose it depends in part on the disease course heterogeneity and the chosen study endpoints, such as pain relief, tumor response, time to progression. For patient advocates (that would be me) a credentialed resource describing this would be useful to have when taking part in IRB deliberations.
Asking after reviewing a meta-analysis pooling RCTs for the efficacy of curcumin as pain relief for arthritis, where selective reporting is mentioned as a limitation.
Not on any major points. We’ll have to agree to disagree on the more minor points about arbitrariness of which variables are collected and which among them are adjusted for. Too many clinical trialists even make an arbitrary decision about adjusting for no covariates vs. adjusting for some.
1 Like
Thanks Erin for the thoughtful reply. The pre-specification issue is separate from my previous points in this thread, in that the points apply whether or not one demands pre-specification.
As I said above, I am fine with the idea of demanding a pre-specified protocol that includes analysis details, such as the list of allowed adjustment covariates and the methods in which they will be used. I say “allowed” because there are statistical methods (such as lasso) that may decide to drop certain covariates based on the data they are given. Use of these methods can be prespecified along with the candidate covariate list (the methods I prefer only shrink the covariate effects instead of dropping some).
Your opening paragraph is a good summary of my view. I’d only add mention of the problem of model reification, which is the prevalent mistake of treating statistical results as if they are properties of reality rather than of the formal mathematical framework (“small world”) from which those results are deduced. “Statistical inferences” are what we get as outputs when the data are compressed into that math framework and transformed by the math algorithm (statistical program) used for “data analysis”. The fragility of the connection of those “inferences” to reality is illustrated by how they can be produced by simply feeding the algorithm made-up (or “simulated”) data. They can also be altered significantly (in any sense) by seemingly small and unrecorded deviations from a trial protocol.
Such deviations may go undetected if only pre-specified analyses are allowed. Even without such deviations there may be good reasons to engage in further unanticipated analyses based on what happened in the trial or what was seen in the data, e.g., nonadherence or drop-out far beyond what was anticipated. I would thus object to banning further analyses that were not pre-specified; they just need to be clearly labeled and justified as such.
As you mentioned, pre-specification is often promoted for avoiding bias. But we know there are threats much deeper than what pre-specification can address, and that are often invisible without analyses beyond anything anticipated at trial inception. Technical statistical issues (like preserving alpha levels) can even be a distraction from such problems, especially In topics with big implications for profit and liability. Consider this recent breaking story:
That kind of story comes in my feed almost daily, including examples of efficacy exaggeration and adverse-event submergence.
On the other hand, you raise a concern about drug safety researchers “crying wolf” and give a storyline which has taken on something of the quality of an urban legend in certain circles. At least, I have seen the story used by industry legal defense teams and their experts to attack researchers who turned out to have actually discovered serious harms, or at worst simply got a false positive (as must sometimes happen in research capable of finding true positives). Often the researchers are accused of data dredging even when the associations in question were clearly pre-specified in the project proposal.
There are of course researchers on both sides who make unfounded claims in courts, media, and paper-mill journals. Nonetheless, while I don’t read them all cover to cover, I have not recognized what you described in the reputable applied journals in my specialty (e.g., Epidemiology, AJE, EJE, IJE, JECH, Annals of Epidemiology). Hence I would ask you to provide some examples involving academic or similarly qualified primary researchers (e.g., NIH funded) who do what you describe and publish it in legitimate peer-reviewed journals. Especially valuable would be an example in which their results were not quickly refuted in a valid manner. I add “in a valid manner” because it seems a common practice to claim refutation based on the “replication attempt” getting p>0.05, yet a direct comparison with the original study displays no meaningful conflict at all, just the expected regression of the effect size toward the null (which the original study could have predicted by providing a shrinkage estimate, as I also recommend).
6 Likes
Dr.Greenland- thanks for your response. I won’t give specific examples of the phenomenon that I call “crying wolf,” since I don’t want to impugn any person’s motivation/reputation specifically. To clarify, my awareness of this phenomenon is not “second-hand news” (or “urban legend,” as you put it) but rather stems from my own experience, over many years, surveilling the medical literature as a job requirement. My comments mostly relate to high profile, mainstream medical journals (e.g., JAMA, BMJ). It’s quite possible (likely, even) that this phenomenon doesn’t manifest to the same extent in the more specialized epidemiology journals you listed. Sadly, though, it’s the mainstream journals that grab the most attention.
In the course of my previous employment, I came to recognize that there are some researchers (granted, perhaps a more vocal but less experienced minority) who craft careers publishing one drug safety-related scare story after another in prominent journals. There is definitely also a subset of researchers/pundits who have gained some level of fame (increasingly common in the era of blogs/social media) by promoting contrarian/conspiracy-minded attitudes toward “the medical establishment” or “Big Pharma.”
To be clear, I’m not saying that most, or even a sizeable proportion of researchers pursue vendetta-based careers. The vast majority are likely true professionals, operating in good faith at all times. But some do appear to pursue a less altruistic agenda. And once it becomes clear to clinical readers that a researcher’s personal ideology affects his decisions about what to study and how, all credibility is lost. I’ll also acknowledge, unreservedly, that pharmaceutical companies sometimes engage in nefarious practices. We’ve seen some doozy drug safety scandals over the years and we’ll see them going forward. They can cause massive harm to patients and to the public’s trust in science. For this reason, the clinical community needs to remain extremely vigilant when examining the design and analytical choices made by companies with massive financial stakes in study outcome.
It would be great if we lived in a world where everyone’s motives were pure and unbiased, but we don’t. Huge financial stakes, the drive to protect professional and organizational reputations, and prospects for personal fame and career advancement can all affect, profoundly, the way that study results are presented.
2 Likes
Related to using pre-specified algorithms like lasso instead of pre-specified models we need to be cautious. lasso seldom chooses the correct variables when there are many candidates, and will not give correct standard errors in the usual sense.
5 Likes
Thanks Erin. Without specific examples of “crying wolf” I can’t say much about that, especially its prevalence and how that compares to its opposite, suppression of concerning evidence; nor can I judge the cases for myself. Regardless, while pre-specification can provide a logical foundation for statistical claims and assurances against bad practices like P-hacking, it provides little practical protection against investigator bias.
One reason is that surveys have found studies claiming pre-specification rarely have their actual activities checked against the pre-specification. Then, when they are checked, serious discrepancies seem common; usually, the authors deny those deviations are of consequence, but others may disagree (especially if they disliked the reported results). A recent highly publicized example is discussed at https://statmodeling.stat.columbia.edu/2024/09/26/whats-the-story-behind-that-paper-by-the-center-for-open-science-team-that-just-got-retracted/
Other reasons pre-specification is not particularly effective against investigator bias is that skilled experts can design a pre-specified protocol that minimizes chances of undesired results and maximizes chances of desired results. In fact, to some extent such design optimization around desirable results is expected for both ethical and practical reasons. For example, RCTs will exclude patients thought to possibly be at high risk of adverse events or unlikely to benefit from the treatment. These exclusions can result in valid estimates of event rates for the exclusive RCT group, but produces underestimates of adverse events and overestimates of benefits in general patient populations.
Less beneficently, certain groups specialize in generating negative studies of harms from environmental and occupational exposures by using pre-specified protocols that seem well powered but aren’t. This can be done by calculating power with no accounting for measurement errors (as is the norm) while using noisy measures of exposure; this ensures low actual power to detect any reasonable effect size. Then of course the resulting p>0.05 is reported as “no association”.
That said, I am in complete accord with your closing paragraph; after all, I have been going on about these “human factors” for many years, e.g., Greenland S, 2012. Transparency and disclosure, neutrality and balance: shared values or just shared words? Journal of Epidemiology and Community Health, 66, 967–970, https://jech.bmj.com/content/66/11/967. But questions remain about concrete ways to deal with the problems. I am all for pre-specification, but it’s worth little if it is not pre-registered and then submitted and published as a key supplement along with the report of results; even then, it can only address a narrow set of biases.
Conversely, absence of pre-specification should not be taken as evidence that a bias occurred (as is often done to attack undesired results). Its absence only indicates that some avenue for bias was left open, which is a source of uncertainty about the results for those who don’t trust that the authors followed good practices.
5 Likes
Sorry just realized I missed this question. The short answer is: it depends ![]() But a good advice from @f2harrell is that it is a good strategy to be a pessimist when designing RCTs. An example is a situation that stumbled a lot of clinicians but kidney cancer patient advocates are well aware of: there were four phase 3 RCTs of adjuvant immunotherapy in kidney cancer (KEYNOTE-564, PROSPER, IMmotion010, and CheckMate-914) of which only one showed a “positive” result leading to FDA approval. Oncologists were struggling across multiple editorials in various clinical journals (Lancet, again Lancet, Nature Reviews Urology, Journal of Clinical Oncology) to explain why the three other phase 3 RCTs were “negative”. There were even discussions that perhaps pembrolizumab is biologically different from nivolumab, explaining the “positive” results of the approved therapy. Which would be extremely surprising. A more plausible and straightforward answer is simple but key differences in the design power of these RCTs.
But a good advice from @f2harrell is that it is a good strategy to be a pessimist when designing RCTs. An example is a situation that stumbled a lot of clinicians but kidney cancer patient advocates are well aware of: there were four phase 3 RCTs of adjuvant immunotherapy in kidney cancer (KEYNOTE-564, PROSPER, IMmotion010, and CheckMate-914) of which only one showed a “positive” result leading to FDA approval. Oncologists were struggling across multiple editorials in various clinical journals (Lancet, again Lancet, Nature Reviews Urology, Journal of Clinical Oncology) to explain why the three other phase 3 RCTs were “negative”. There were even discussions that perhaps pembrolizumab is biologically different from nivolumab, explaining the “positive” results of the approved therapy. Which would be extremely surprising. A more plausible and straightforward answer is simple but key differences in the design power of these RCTs.
As we subsequently discussed in two editorials here and here, the trick when interpreting these RCT results is to not dichotomize them into “positive” vs “negative”. This will help notice that the “positive” KEYNOTE-564 RCT had higher signal-to-noise ratio while the other three RCTs were more noisy and imprecise albeit still pointing in the same direction. This is because Merck (as they often strategically do for RCTs they care about) aimed for a 95% power for KEYNOTE-564. Notice in the Table from our European Urology editorial (“EP” is estimated design power) that the other three “negative” RCTs were designed to enroll fewer patients in the adjuvant setting compared to the corresponding metastatic setting. Which is bogus since the metastatic setting has far more common “events” than the adjuvant setting. The KEYNOTE RCTs were the only ones that aimed to enroll more patients in the adjuvant setting than the metastatic one (150 more patients to be precise):
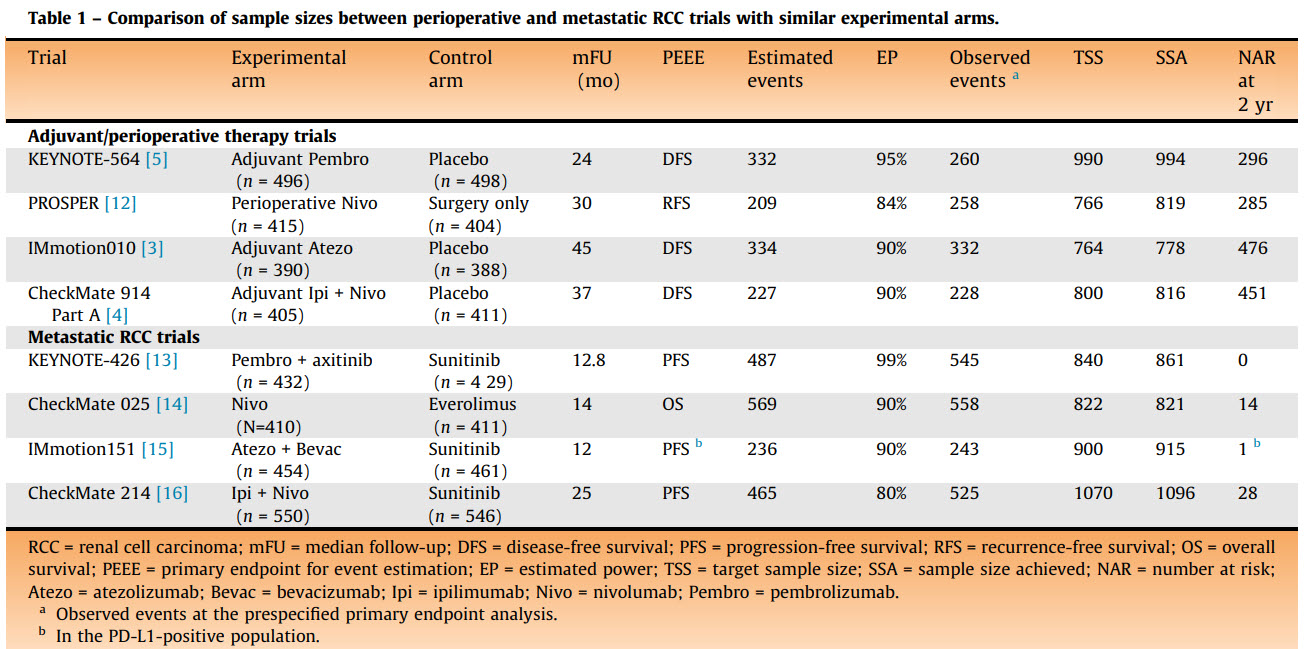
Having said that, our empirical data show that the median actual power of phase 3 RCTs in oncology is only 49%, which nevertheless is far better than the median actual power of 13% achieved by RCTs across medicine catalogued by the Cochrane Database of Systematic Reviews to guide clinical practice. But it still falls short of the prestudy “planned” power of ≥80% that most oncology RCTs aim for. This adds to the argument in favor of being pessimistic when designing RCTs.
Note also that, as recommended by @Sander above, we have made a free calculator based on these data that allows anyone to easily generate data-driven shrinkage estimates from any phase 3 RCT in oncology by simply entering their standard reported hazard ratio and 95% CI. In an upcoming study (draft version of its calculator here) we aim to estimate the replicability of phase 3 RCTs in oncology, defined as the probability of obtaining a P ≤ 0.05 in a replicate RCT with the same direction of effects as the original phase 3 RCT.
2 Likes
We need to find a way to further publicize this entire topic. There is so much value here, and these discussions need to make their way into biostatistics, epidemiology, and clinical trials classrooms.
4 Likes
From a provider/patient perspective, how does one interpret an adjusted treatment effect? In other words, if someone goes to a provider wondering whether to start a statin, but the statin’s treatment effect in the literature has been adjusted for autoimmune disease, what does the conversation look like?
If there is no interaction between treatment and presence of autoimmune disease, then it’s easy: the treatment effect is estimating would would happen had a patient with autoimmune status x been given treatment B vs. treatment A, for any x.
1 Like
One clinical trials teacher has just been trying to figure out how to work it into her course this semester ![]()
3 Likes