Deletion of this post pending
Deleted post pending
2 Likes
Lawrence I’m going to delete these last two replies as they belong in one of the topics you have already created and are not related enough to the current discussion.
Ed I’m glad you are engaged here, and that you received two complete answers to your concerns. Your point of view is one of the most common misconceptions of clinicians, and if someone as learned as you can hold that you can help us come up with optimum educational strategies for clinical audiences. In addition to what was said, @Stephen points out that the inclusion/exclusion criteria just provide bounds for enrollment but do not at all dictate the final distributions of patients who enroll, which may be narrower than the inclusion criteria.
Ed your statement would be correct if you modified it to
RCTs only reliably inform clinicians about treatment effect of an intervention in the subset of patients for whom variables meaningfully interacting with treatment (but were forgotten to be included in the primary analysis as interaction terms) have distributions in clinical practice that strongly overlap with distributions in randomized patients.
3 Likes
As an additional clarification, the covariate distribution of patients we see in clinic and for whom a therapy may be appropriate may be narrower for some covariates and wider for other covariates compared with the patient sample enrolled in the trial that established that therapy. An easy example is the covariate “time period” which will almost always differ for future patients treated after the trial was completed.
This is why representative sampling from populations is typically far less important for transportability purposes than representativeness of causal mechanisms. If a therapy acts through a mechanism that my patient in clinic lacks then this therapy is inappropriate even if the patient otherwise perfectly corresponds to the trial sample.
4 Likes
This effort is highly commendable. I will offer a few critiques/comments. The term “joint intervention” is opaque to me. While a selection process (S = 1) is clearly implied, its role is fundamentally different from that of an intervention and IMO should not be treated as such. Selection defines the causal domain within which randomization identifies an effect; it is not itself an intervention on the system. Moreover, because S = 1 often functions as a gate that aggregates heterogeneous causal structures, it is frequently the element that undermines and indeed preempts transportability rather than enables it.
One approach might be to show which RCT designs render transportable results and which do not (and why). Identifying the potential effect of using surrogates, as in the original FDA Flecainide trial that despite being internally valid, when transported rendered a NNK (number to kill) of about 20 in 20 months and was reversed by the highly transportable RCT, CAST.
My point is that a discussion about transportability vs generalizability will best advance the field not by showing the distinction is artifactual (which is largely a internal semantic issue among the erudite) but rather by pointing out that the integrity of transportability is the value component for the public. As it was with the pre CAST trial, Internal validity is a pitfall likely to cause a false sense of safe transportability and harm if the results cannot in reality, be generalized/transported without causing harm. So a paper which defines the quality checks relevant RCT transportability would be a landmark.
I don’t follow this point perhaps because IMO the term “pragmatic trial” does not rise to the level necessary for scientific discourse. One cannot seek to distinguish between terms like generalizability and transportability when using nebulous terms like “pragmatic trial”. This may be my weakness so please explain what you mean by a “pragmatic trial” in more precise causal language (eg do-calculus).
Very interesting perspectives @Alessandro_Rovetta. However I take a slightly different view. Lets step back and call the big picture “external validity”, then under this we have two aspects - generalizability and applicability.
Generalisability applies to descriptive studies and surveys rather than analytical studies and refers to inference of overall sample statistics back to a target population – which is not possible from non-probability samples. Therefore, when the statistic of interest is an overall sample statistic (e.g., prevalence, mean value for the whole sample), the non-probability sample estimates a quantity for its own source population rather than for the general population.
Applicability refers to the ability to use the difference in outcomes between treatment groups in analytical studies for decision making in health care settings. There is no requirement for samples to be selected randomly from the population, as the expected difference between groups might be biased regardless of sampling strategy. Corrective actions to ensure applicability include random allocation (randomized trial) or matching, weighting, regression adjustment, etc. in observational studies. The corrective measures are needed for the random variables expectations to line up with the causal contrast we want. This is what we mean by internal validity and such internal validity is a pre-requisite for applicability. A trial with good internal validity might have a sample that may not be representative of those subsets of the populations to whom the results would usually be applied, but there would still be applicability of these results to everyone outside the study subset if those subset variables do not lead to heterogeneity of treatment effects. If there is heterogeneity of treatment effects, then consideration is needed of applicability beyond eligibility criteria.
Applicability concerns whether study findings are relevant for a target setting, whereas transportability addresses whether and how causal effect estimates can be formally re-estimated for that setting; the former does not require the latter, but in my view the former subsumes the latter. Therefore I do not think your paper should be talking about generalizability vs transportability - rather it should be talking about generalizability vs applicability.
Finally, transportability and portability should be distinguished. Portability across contexts depends on if an effect measure has variation dependence or not and is distinct from transportability.
2 Likes
I just read your paper and you concluded that “Ensuring representativeness does not mean finding a target population that demographically mirrors the trial sample (representative sampling); rather, it means verifying that the target setting sufficiently preserves the causal attributes, the biological mechanisms, and the environmental conditions required for the intervention under study to exert its effect (representative causal mechanisms).“
I think that ensuring representativeness DOES indeed mean finding a target population that demographically mirrors the trial sample (representative sampling) - aka generalizability and applies to descriptive studies and surveys. We should not strive for generalizability or representativeness in analytical studies. Instead, within the latter, the process of verifying that the target setting sufficiently preserves the causal attributes, the biological mechanisms, and the environmental conditions required for the intervention under study to exert its effect (representative causal mechanisms) is what applicability as well as transportability allude to today. Both require internal validity as a prerequisite but transportability answers one specific sub-question inside the broader question of applicability. I guess causal inference needed a way to say that assuming the study is internally valid, assuming the intervention is well-defined, assuming outcomes are comparable, then let us isolate only the problem of moving causal effects across populations and have a claim that can be proven false under a causal model. Applicability moves beyond this to say that assuming internal validity and the right effect measure under the right causal model, is the intervention relevant and feasible beyond eligibility criteria, are outcomes meaningful and will use change decisions?
You might find the paper I mentioned above interesting. It addresses some of your points, e.g., footnote 11.
Findley et al. [17] make a strong distinction between “generalization” in which results reflect the population from which a sample was drawn, from “transportability” in which results travel to a new population. The distinction is not helpful, however, since generalized claims always must transport across time, and often space, to be meaningful (as argued in Munger [31]).
and the more provocative statement:
Causal specification also calls into question any assertion that internal validity has priority over construct or external validity. Such a claim dates to Campbell [68, p. 297] who stated internal validity as a “basic minimum” for science, and when comparing internal to external validity, he writes, “ Internal validity is the prior and indispensable consideration.” For a more recent statement, see Guala [69, p. 1198]. The reasoning holds that if an experiment is confounded, nothing can be learned from it, and hence, internal validity has a lexical priority over considerations regarding the other types of validity.
However, in the present framework, deductive validity does not require that the validities must be considered in any specific sequence or ranked in their priorities, or that internal validity must be present before one can consider the other types of validity. For example, it is unclear how one can say that knowing an internally valid (directional, unconfounded) cause occurred must precede knowing what was the cause and what was the effect (i.e., construct validity). Similarly, it is also unclear how one can say knowing an internally valid cause occurred can precede knowing whether the setting has the necessary conditions to enable the cause (i.e., external validity). Because all three validities are necessary, no causal generalization can be successfully deduced without all three. None stands separate and prior.
2 Likes
I think a major problem here is language usage. Clinicians and statisticians use very different terminology and that used by statisticians is hard to understand by clinicians. We may be talking about the same thing, but the clinicians may not understand. Admittedly, I am confused by some of this.
Here are scenarios I have experienced clinically that motivated my reaction to this discussion about RCT portability and generalizability:
- Very old women with cognitive limitations gets chemotherapy for breast cancer c/w results for those treatments from RCTs. Toxicity from the drugs is much more severe than predicted from the trials because of the patients age (olderwomen excluded from the trials). The patient becomes severely demented because of the chemo (which, because it was not tolerated had no effect on the tumor). Rapid pregression to dementia is not a factor in the RCTs because the women included in those trials were too young to be at risk for this complication. I have seen this happen many times in clinical practice.
A very elderly patient with HTN is treated with drugs given at doses derived from RCTs. These elderly patients are much more prone to hypotension from the drugs. They may fall from the hypotension and be at more risk for severe injury from the falls because of their age. The treatment benefit from stroke prevention may be about the same for the elderly and young but the benefits from reduced risk from cardiovascular disease are not present in the elderly.
- I am looking at a very large database of GLP-1 RA treatment for obesity. Only about 1% of patients get the dose and duration of drug shown to have large effects on weight loss in RCTs. 80% of patients treated with the drug have to stop taking it in the time period (with 18 months of drug initiation) included in the RCTs of this drug. How do the results of the RCTs for these drugs apply to the universe of obese patients?
It would be helpful to translate how treatment interactions of covariate distributions apply to these scenarios. If you can do that, your ideas will have much more influence on clinicians and clinical medicine.
As an aside-communicating difficult statistical concepts that appear in medical research to clinicians was a major goal of the JAMA Guide to Statistics and Methods when we developed it. Clinicians loved those articles. They were also highly cited.
Ed
6 Likes
I shall give scenario 1 a try.
To simplify, assume that chemotherapy affects cognitive ability similarly for all involved patients, on a relative scale. For example, the chemotherapy might increase the odds of delirium by a factor of 3 (OR = 3).
Younger patients, have a low baseline risk for delirium, let’s say 1%. Even though the chemotherapy increases their baseline risk, their overall risk for an episode of delirium is still pretty close to 1\% (\sim2.9\%).
Older patients, with many comorbidities might have a high baseline-risk, let’s assume 10\%. And OR of 3 would increase this from 10\% to 25\%. The absolute increase in risk is much higher for older people, even though the relative increase is the same.
We should not look at the average rate of delirium in the RCT and just assume that this applies to our patient. Of course it won’t, it also did not apply to patients in the trial. We should use the RCT to estimate a relative effect, which we can then combine with the baseline risk of the patient in front of us, to guide our decision. If our patient population in the RCT is so young / healthy that the AE virtually never happens, then we have a problem, because we can’t estimate a relative increase in the odds of the side effects.The baseline risk of the patient might be estimate through larger (even observational databases) or just be based on clinical experience.
1. Younger Patients (Baseline Risk: 1%)
Odds_{baseline} = \frac{0.01}{1 - 0.01} = \frac{0.01}{0.99} \approx 0.0101
Odds_{treatment} = 0.0101 \times 3 \approx 0.0303
Risk_{treatment} = \frac{0.0303}{1 + 0.0303} = \frac{0.0303}{1.0303} \approx 0.0294
- New Risk: \approx 2.9%
- Absolute Risk Increase: 2.9\% - 1\% = 1.9% points
2. Older Patients (Baseline Risk: 10%)
Odds_{baseline} = \frac{0.10}{1 - 0.10} = \frac{0.10}{0.90} = \frac{1}{9} \approx 0.1111
Odds_{treatment} = \frac{1}{9} \times 3 = \frac{3}{9} = \frac{1}{3} \approx 0.3333
Risk_{treatment} = \frac{1/3}{1 + 1/3} = \frac{1/3}{4/3} = \frac{1}{4} = 0.25
- New Risk: 25%
- Absolute Risk Increase: 25\% - 10\% = 15% points
3 Likes
These are great clinical examples. Fully agree that a huge reason why bad statistical practice remains so prevalent is failure to effectively translate complex statistical concepts into language clinicians will understand. This is the “rate-limiting” step in fixing the problem, yet very few in the statistical community seem to prioritize it. As a result, clinicians are left trying to do the translating themselves, often with very mixed results. But who fields the subsequent criticism? Clinicians.
For example (and stipulating that I appreciate the response), everything after the third bullet point in post #28 of this related thread went right over my (clinician’s) head: Critique of paper on generalizability of oncology trials
3 Likes
I think we can come up with simple language when isolating a primary efficacy outcome from everything else (e.g., not dealing with your hypotension example). If “pure” efficacy interacted only with sex, efficacy results from the RCT would not transport to either a male or a female if sex and sex \times treatment interaction were not included in the primary model and if the interaction effect is substantial.
The situation you wrote about is a case of multiple outcomes. You may or may not want to call some of the outcomes efficacy outcomes and some of them safety outcomes. Then your example boils down to differential effects on safety outcomes by age, even if age does not modify the primary efficacy benefit. Now the problem has come that outcomes are multi-dimensional (multivariate even at a single follow-up time) when we insist on analyzing the components separately. We need to quit doing that.
As a modeler I would work with clinical colleagues to specify a model that reflects what is going on. This model would be specified to create the ability to tell which treatment gives patients better outcomes, i.e., redistributes the severity of outcomes towards the good end of the outcome spectrum. This relies on our ability to count things such as inability of tolerate treatment, toxicity, and other side effects as bad outcomes, just not as bad as cancer recurrence or death. One general way to do this is the have an ordinal longitudinal outcome where for each time period (e.g., week) we code the patient status according to the worst thing happening to the patient in that period. From that a Wilcoxon test-like analysis will tell us which treatment patients fare better on, even though the model is misspecified (by say assuming proportional odds, i.e., constant treatment effect over outcome levels) and thus does not provide accurate estimates of treatment benefit for specific parts of the outcome spectrum. To get accurate estimates we have a “treatment by outcome interaction”, i.e., allow for special effects of treatment for safety outcomes that disconnects them from treatment effect on primary efficacy outcomes.
In that formulation, there is no single parameter that captures treatment benefit. But with repeated measures, magic happens in a way that allows one to compute clinical readouts that are relevant to patient decision making. Define one or more zones of good outcomes. For example Y < 5 might indicate absence of clinical events and significant toxicity and patient is at home. Then we estimate the covariate-specific mean time in a Y < 5 state for each treatment and the difference in mean times. The result is e.g. expected time gained for patients being in a good state. This averages over the treatment \times Y interactions in a reasonable one, creating a one degree of freedom contrast with excellent power.
This situation is harder to describe for clinicians but I’m sure we could all come up with excellent language for non-statisticians.
3 Likes
Here is an example clinical review on pragmatic trials. For a do-calculus description of a key feature of pragmatic trials, take the selection diagram of figure 6 here (which, for further elaboration, corresponds to figure 6 of this selection diagram-based RCT review):
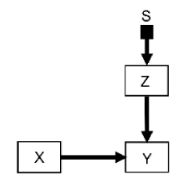
Where S is the selection node, Z is a vector of all patient covariates, X is the treatment choices under investigation and Y is the outcome of interest. Let S = 1 denote the trial population and setting, and S = 0 denote the target clinical population and setting. The key pragmatic trial condition is that the trial recruits a representative sample so that P(Z | S=1) = P(Z | S=0). And therefore: P(Y | do(X), S=1) = P(Y | do (X), S=0).
In the notation of Bareinboim and Pearl’s classic work (which is the key inspiration behind our causal/selection diagram reviews linked throughout this thread) the selection node S is d-separated from Y and thus the transport formula is the simple identity P(Y | do(X), S=1). This is in contrast to standard “non-pragmatic” trials whereby the transport formula would be:
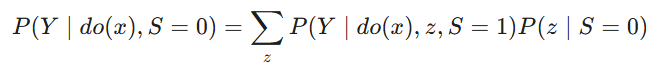
i.e., the causal effect in the target population (S=0) is the causal effect in the trial population (S=1) re-weighted by the distribution of covariates Z in the target population.
@Johannes_Schwenke 's correct intuition is that this transport formula simplicity often masks a statistical penalty: pragmatic trials are inherently “noisier” and the treatment signal is often “diluted.” In statistical terms, efficiency is usually defined by the variance of the estimator or the sample size required to achieve a certain power. There are at least two reasons why pragmatic trials can have lower efficiency:
-
Variance inflation (heterogeneity penalty): Standard trials try to artificially reduce noise in part by selecting more homogeneous populations, i.e., truncating the distribution of Z making the variance of Z small. Pragmatic trials accept the full “real world” distribution maximizing the distribution of Z, and consequently its variance.
-
Compliance penalty: I have only focused above on the key intuitive advantage of pragmatic trials which is the potential simplicity of its transport formula, which carries a hidden cost. A key potential disadvantage, intuitive to clinicians and methodologists alike, is that pragmatic trials may often have lower compliance with the treatment strategy X. That obviously can add further noise in addition to the variance inflation above. Our causal inference expert clinician / methodologist Kerollos Wanis has been thinking extensively about these adherence topics, e.g., here and here.
1 Like
Correct me if I’m wrong but if If one defines pragmatic trials by P(Z |S=1) = P(Z | S=0), then one has not defined a class of trials, but rather a limiting case in which transportability is trivial if S=1 is a valid gate.
My experience with pragmatic trials is that they often have a broad (pragmatic) S=1 which results in a high U rather than resolving transport. From a causal perspective, this is not a semantic distinction because a widening of inclusion without mechanism isolation replaces visible exclusion with invisible causal mixing.
At that point any discussion of transportability vs generalizability becomes superfluous.
I appreciate the intent behind pragmatic designs, and I suspect there is much we agree on. Since this is tangential to the instant discussion, I’d be happy to continue the discussion offline if you’re open to it
2 Likes
Just to clarify: while I am pretty sure one group or another will tap me into advising / supporting pragmatic designs (and will be more than happy to help them as needed), they are not my natural inclination and I find more limitations than benefits in many such designs. Been saying that for years and fully aware how hard it is to see for many stakeholders who believe that pragmatic trials are always preferable. Grateful to this thread for allowing a comprehensive elaboration on these topics.
2 Likes
Where I think pragmatic trials shine is when there are two widely-used competing clinical practices, such as the cluster randomized trial we did on saline vs Ringer’s lactate IVs at Vanderbilt on 10,000 patients.
5 Likes
How about this attempt at translating the paper’s messages for clinicians?
RCTs aim to optimize benefit and minimize risk to their enrolled subjects, such that new therapies have the best chance of getting approved by a regulator. Approval signifies that:
-
the therapy has demonstrated intrinsic efficacy under the conditions present in the trial (whether that efficacy is clinically important is another issue). Subjects in the treatment arm had better outcomes, as a group, than subjects in the placebo/active comparator arm, with regard to the primary outcome of interest; and
-
the risk/benefit profile for the therapy, under the conditions present in the trial, appeared positive at a group level (one arm vs the other). There were no safety signals concerning enough to hinder the therapy’s approval.
Suggested steps for deciding whether to apply an approved therapy to a patient in the postmarket setting:
Step 1: Assess whether the group-level efficacy signal and important contextual factors (e.g., monitoring/supportive care) can be “transported” from the pivotal RCT to the individual patient’s context. This is a biologically- and situationally- informed decision. The patient’s disease must share those biological features which mediated the therapy’s group-level effect in the pivotal RCTs. Contextual features that might have facilitated detection of the therapy’s group-level efficacy and minimized its harms (e.g., monitoring, supportive care) must also be transportable to the patient.
Step 2: Consider whether the risk/benefit profile of the therapy is likely to be favourable for the individual patient. This step is much more complex than Step 1; the patient’s unique covariate profile/circumstances must be considered. Clinical expertise and judgment are required.
The risk/benefit calculus for a therapy in individual patients can sometimes depend heavily on the patient’s covariate profile (e.g., age, list of comorbidities, concomitant medications). Yet only a small portion of the available “covariate space” defined by a trial’s inclusion criteria might actually be filled by patients who end up enrolling in the trial. It follows that we can’t assume that a favourable group-level risk/benefit profile for the therapy among highly-selected trial subjects will be transportable to individual patients following approval. Conversely, provided that a patient in the postmarket setting possesses the necessary biological drivers of therapeutic efficacy, he should not be denied therapy simply because he is “too dissimilar” to patients in the trial with regard to other covariates. His risk/benefit calculus will not necessarily be unfavourable just because he doesn’t resemble trial patients “closely enough.”
For these reasons, clinicians should not compare the covariate profile of individual patients in the postmarket setting to the profile of trial patients when trying to decide whom to treat. Provided that biologic drivers of therapeutic efficacy are expected to be shared, the degree of overlap between the covariate profiles of trial subjects and those of individual postmarket patients will simply make extrapolation of the risk/benefit profile more tenuous.
4 Likes
This schematic might help. Note I have renamed “transportability“ to “causal applicability“
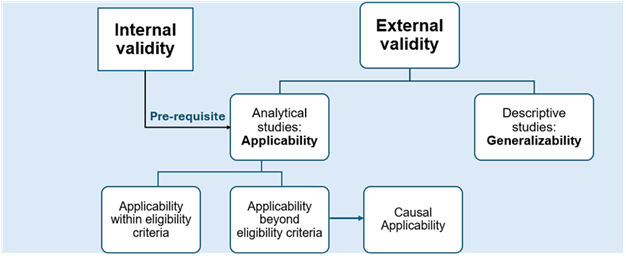
This is from a paper we are writing titled “Redefining external validity”
Looks quite interesting but I don’t follow it completely. To me the missing piece is causal object explication . Eligibility criteria is an opaque term. What is it? This is where the fundamental error of communication between stat and pathophysiology expert occurs.
P(Y | do(T), E) what does that even mean? It could easily be anything and often is.
For example it could be
E = {C1, C2, C3, …} or E=g(symptoms, thresholds)
Where: g is the eligibility rule based on non-cause specific clinical findings of thresholds.
Please provide supportive explanation. Particularly of E.
Btw “Causal applicability” is an excellent term. There is a little expert heuristics in that function, I suspect. IMHO the graph is powerful but without causal explication it the graph is easily misinterpreted.
1 Like