Over on X there is a debate relevant causal inference vs RCT design. But what is an RCT?
Since Bradford Hill’s landmark study we have seen the RCT morph into two species.
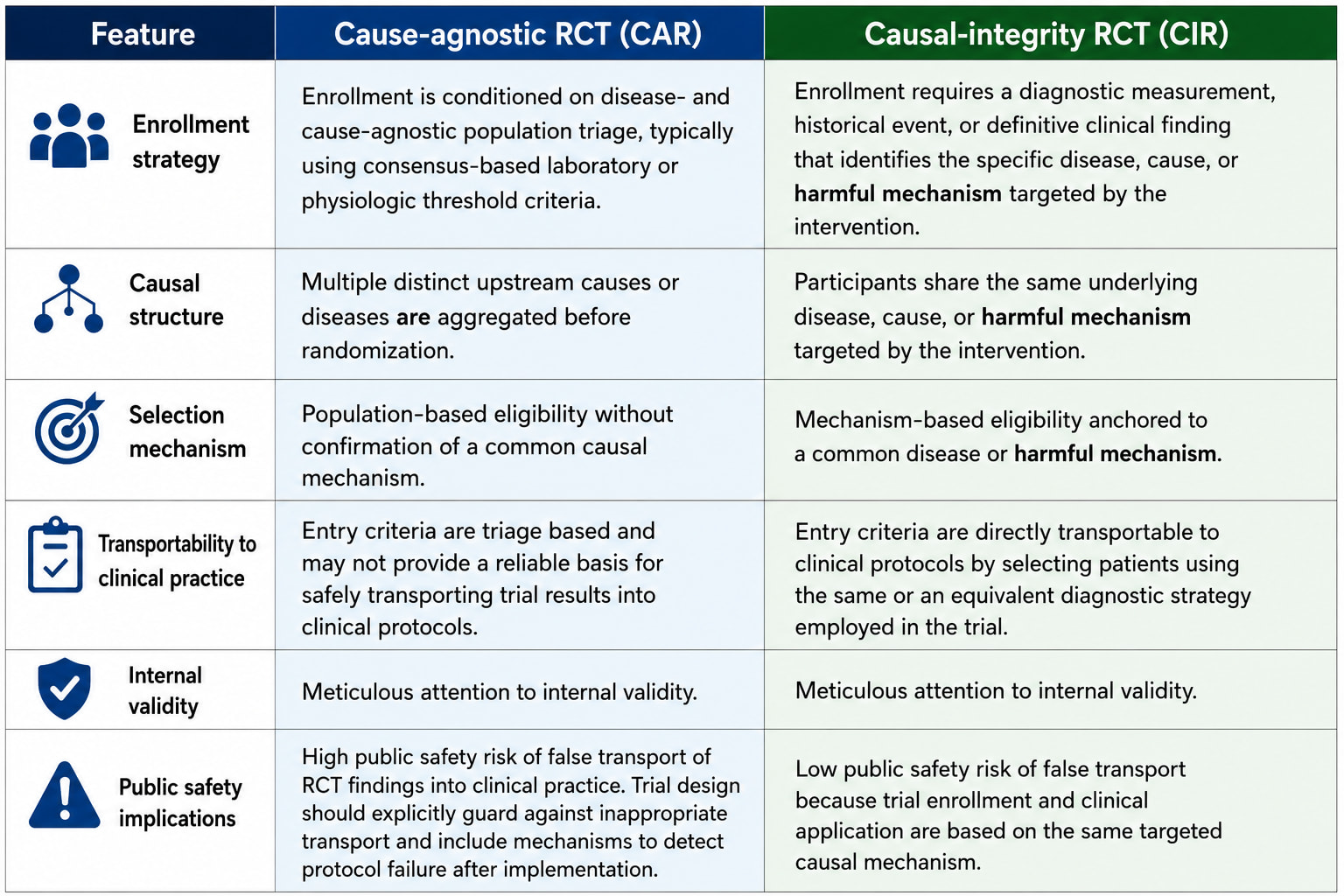
So can we no longer speak of or debate RCTs as if they comprise a unified entity… a thing. There are CIRs (causal-integrity RCTs) for example the original Bradford Hill causal design, and CARs (causa-agnostic RCTs).
-
a CIR conditions on a cause AND a covariate vector.
-
a CAR conditions one or more thresholds (S=1) and are agnostic to cause.
The RCTs of synthetic syndromes like sepsis and ARDS are CARs.
Slide showing the left side of the DAG of a CIR (top panel) in relation to a CAR with the emphasis that CARs are generally NOT on the causal path so they generally crash. .
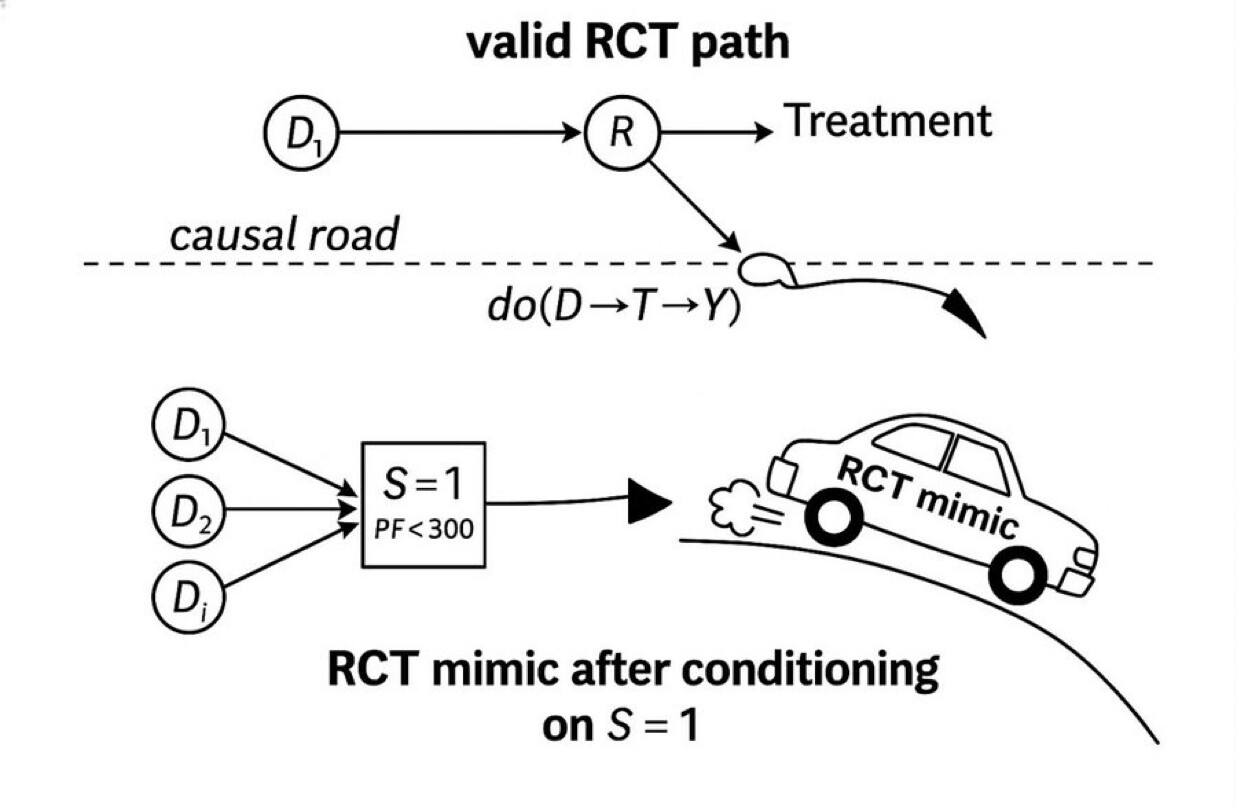
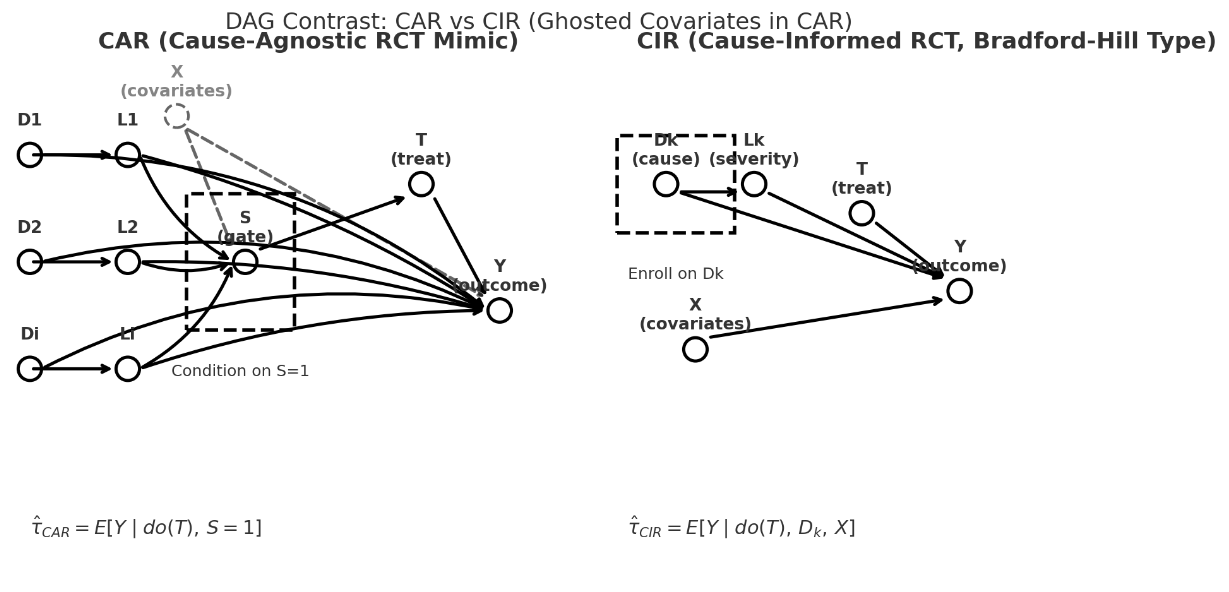
CIRs are what we all thought RCTs are. They have morphed into the much easier CAR which use non-disease (and non-cause) specific threshold sets as triage thresholds as the gate making case finding and a high n easy but there is no causal unity so they are internally valid (as a function of randomization) but externally non transportable.
The first thing a clinician should ask is: is this study a CIR of a CAR?
This is not to say that CARs (cause-agnostic RCT) are always invalid. Covariate matching might be valid if the same matching can then be applied to the population external to the CAR but not if conditioned on a threshold with a few perceived covariates as in the classic Petty Bone RCTs, the invalid CARs (RCT mimics) which have devastated the image of the RCT in general in critical care.
This “covariate” matching is potentially accomplishable by computer based subphenotype relational time pattern recognition.
Success of such an advanced covariate based CAR technique is likely capturing the unknown cause or mediator in a sense converting it into a CIR equivalent.
Note that matching “covariate” vectors by prognosis (eg AUC-mortality) cannot accomplish this potential CAR to CIR conversion to equivalency.
So the critical care clinicians have to lead the way because these causal paths and covariate vectors and trajectories are complex.
this is a call for CI and design community “synergy,” This distinction of CIR and CAR complements Pearl’s formal identifiability conditions by applying them to the clinical gate itself and it honors the design focused axiom that; “the integrity of the design determines transportability.”
Yet If the CI school insists that the design world must BEGIN by accepting layered propensity-weighting assumptions as definitive, and the RCT design establishment continues to ignore the categorical difference between a Bradford Hill-style CIR and a modern Petty-Bone CAR, then we will remain trapped in the epistemic interface gap.
If this lack of collaboration persists, it is up to clinicians can break this impasse because trialists have been unable to engage in such antidogmatic discussions. Witness you saw all of them run away here. The present state is creating disciplinary dissonance because it challenges 5 decades of methodological consensus. Indeed this analysis questions the heart of the present and decades old hierarchical critical care task force controlled syndrome science itself.
Of course the intellectual antipathy is palpable between CI and RCT design communities. You can cut it with a knife and it has long been unrelenting. I don’t see any intellectual sympathy coming from either camp. So this time it’s up to the clinicians to take the initial steps to save their patients from the perils of intractably siloed epistemic orthodoxy.
Firstly: by telling the CI community we yet don’t know enough to START with layered assumptions and,
Secondly: by refusing to consume Petty-Bone CAR based research and rejecting the task-force-driven scaffolds upon which CAR “science” rests.
I look forward to further discussions.