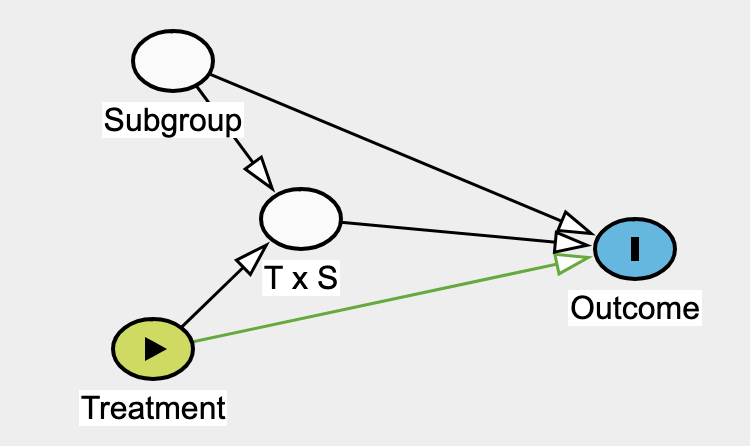
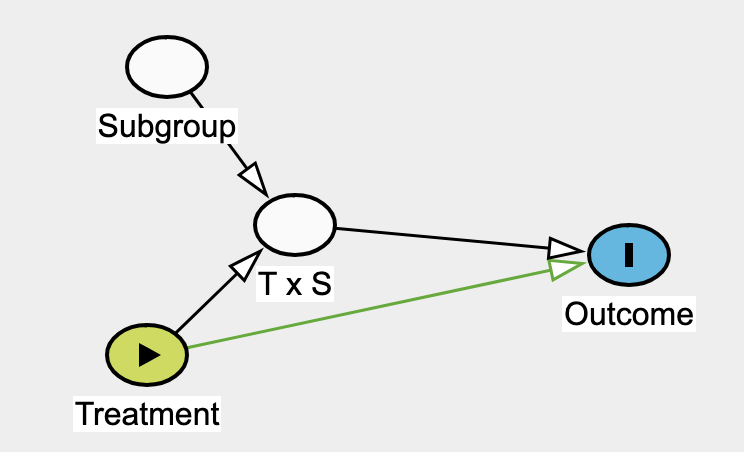
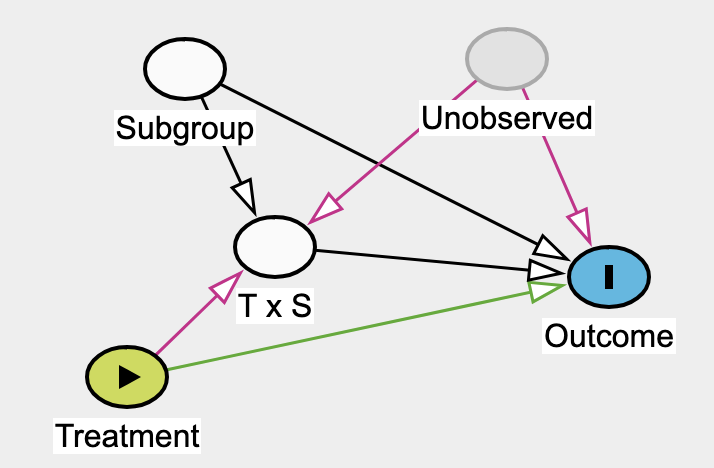
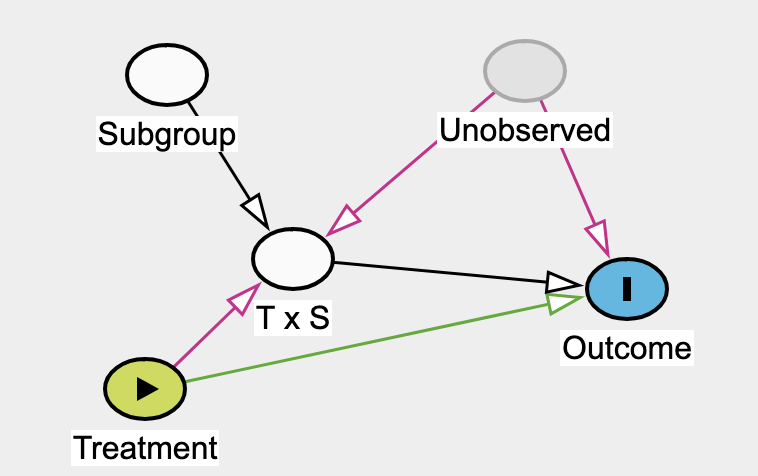
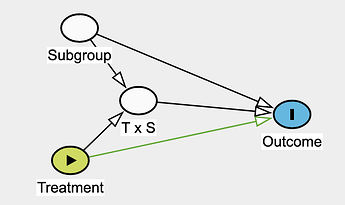
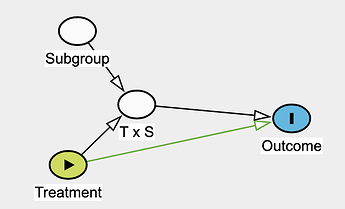
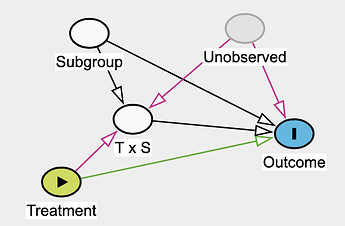
Effect modifier: they suggest reserving this strictly for situations where the second factor (the modifier) does not have a direct causal effect on the outcome itself.
I want to extend this framework to subgroup analysis in randomized trials, where one would focus on the parameter related to T \times S to answer the question:
Is the treatment effect different between subgroups?
I noticed that stratifying by subgroup and including the T \times S parameter could open a backdoor path in both interaction or effect modifier frameworks if an unobserved variable influences both T \times S and the outcome:
Has this phenomenon in subgroup analyses been described before in RCTs? How should one interpret such analyses if there is a risk of opening backdoor paths through unobserved variables?
Yup, this remarkably came out at the exact same time we described the same thing using similar DAGs (a major difference is that we adapted them towards selection diagrams and described the do-calculus considerations). See also here.
The potential collider bias you point out is what @Stephen eloquently refers to when cautioning that in subgroup analyses we can assign individuals their treatment levels but not their covariates.
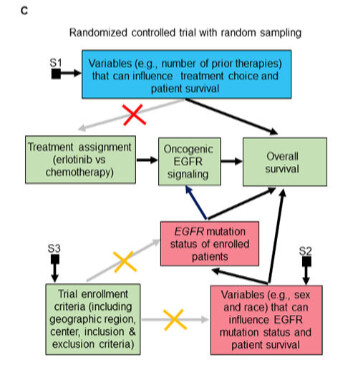
This was extremely helpful! I have a clarification question: In the example you give, if EGFR mutation was the only cause of oncogenic EGFR, would we still get a biased estimate in a subgroup of patients with oncogenic EGFR signaling? I would think not as in this instance this would be equivalent to an RCT in patients with EGFR mutation, would it not?
I’m also somewhat confused by the treatment indicator causing EGFR signaling. Temporally, I would assume EGFR signaling to already be present / absent before randomization (?) .
This is exactly the kind of question that will help me in figuring this out. It seems to me that if you could have done a meaningful and easily interpretable clinical trial on the subgroup in question, you should be able to figure out interaction effects involving that factor in a larger trial.
People who are part of the subgroup also have a higher/lower risk for the outcome than people not part of this subgroup, even if they are not given the treatment. That’s @Pavlos_Msaouel EGFR example. People from the subgroup also have a different relative treatment effect (on the scale of interest) when given treatment than people not in the subgroup.
Why is there an arrow from \text{Unobserved} \rightarrow \text{T x S} ? We are not modeling the \text{T x S} Interaction using U?
I think this corresponds to this graph from Pavlos publication. But the above DAG would somehow lead me to think that people who are part of U and \text{subgroup} have a different relative treatment effect than people who are part of \text{subgroup} and not U?
Quote from Epi paper which helps a bit:
It is possible that there are factors that influence the likelihood of both expo- sures occurring concurrently (marked by an arrow into the interaction E G node) and also influence disease risk (marked by an arrow into the disease D node); such a fac- tor would create a back-door path and this would be ex- plicitly visualized in the DAG (see Figure 2c). Continuing with our example of smoking (E), asbestos (G) and lung cancer (D), a potential confounder (C) would be a factor that increases the likelihood of both smoking and asbesto- sis exposure, such as socio-economic status. Although this back-door path could also be captured by arrows from C to both E and G if the E G node were omitted, the inter- action node prompts the researcher to think about factors that affect both exposures simultaneously.
Yeah, this why I chose to model the confounding influence of U on the baseline variables that we know at time 0. While simpler than modeling the interaction, it has very high payoff in practice to focus on this.
Correct. This is also related to @f2harrell’s comment:
Indeed, we exactly discuss and formalize this point in Section 3.4 here using the example of HER2. Notice that contextual knowledge from correlative and functional lab research is needed to choose the subgroup and develop the therapy for it. Hence the focus on that paper on transporting such knowledge across domains.
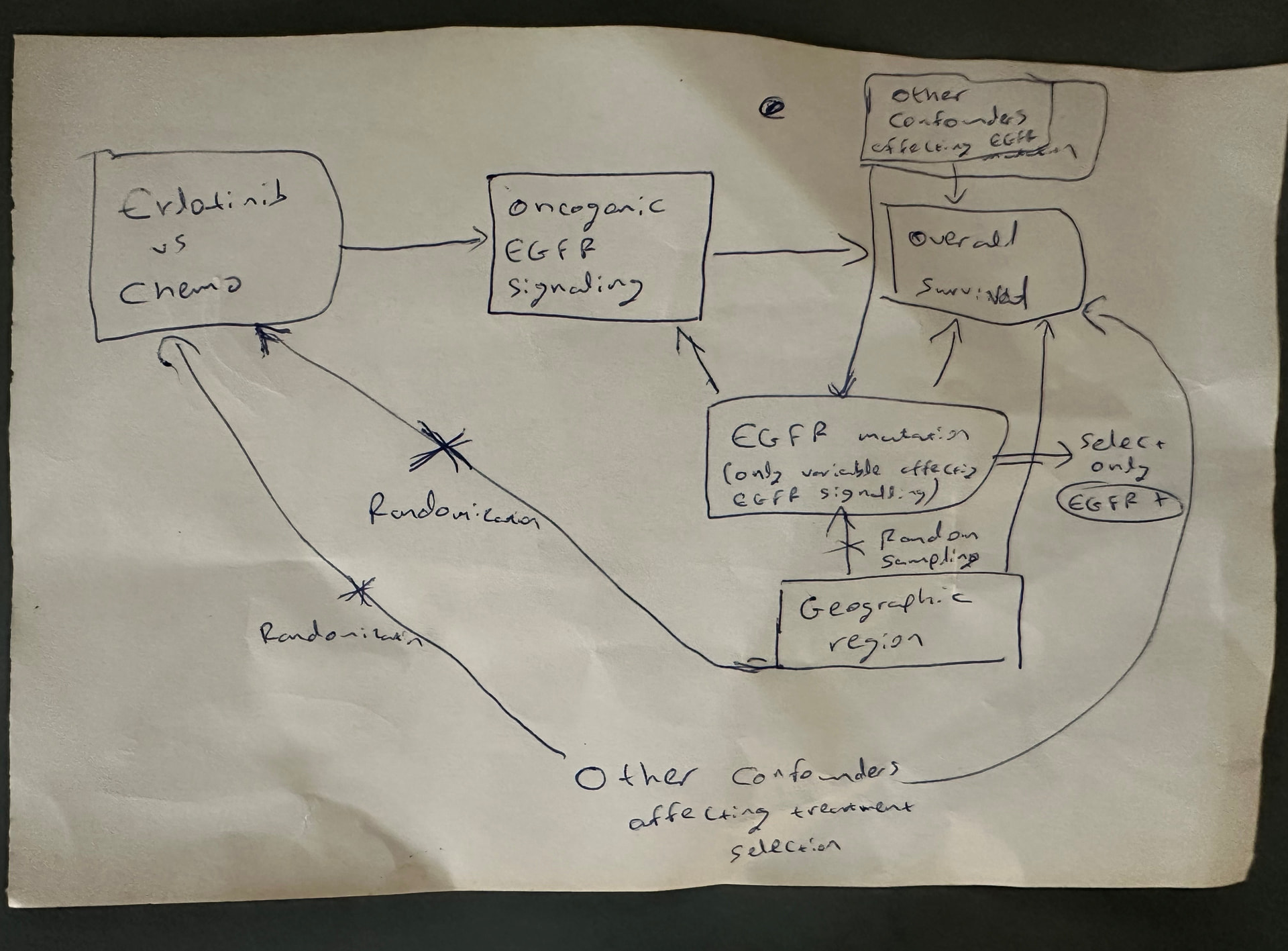
Notice the qualifier oncogenic EGFR signaling (not just all EGFR signaling which exists in normal cells). The oncogenic mutations on the tyrosine kinase domain of EGFR induce oncogenic EGFR signaling that can then be targeted (causally modified) by EGFR tyrosine kinase inhibitors.
I had forgotten that the Impervious to Randomness paper focused on teasing out oncogenic EGFR signaling. The DAG was drawn in my head during a day hike with my then soon-to-be wife around Santorini on 7/18/2021. I drew it on the piece of paper below and then wrote that manuscript as a way of not forgetting this concept. But as shown here (from 1:34:00 onwards) that EGFR pathway mental dissection allowed us subsequently (in May 2022) to come up with the most powerful therapy developed to date for renal medullary carcinoma – the deadliest kidney cancer in adolescents and adults. There are patients alive today (some even cancer free) that would otherwise no longer be with us if not for this.
Once we started thinking in a structured way about randomizing a patient’s covariates to remove these confounders this led to sampling theory. Then we spent a lot of time thinking about the implications of random sampling versus random treatment assignment and wrote this very long paper to summarize these points.
Depressingly, this line of thinking then allowed me to recognize the oxymoronic nature of randomized non-comparative trials (RNCTs). To this day, I struggle to convince some biostatisticians why RNCTs are such a bad idea. These DAGs are one method of communicating these concepts but they still need attention and may not work for everyone. Different tools may be a better fit for at least some people.
This was an opinion paper in IJE. I have decided to write a counter-opinion given so many (in my view ambiguous) attempts at inserting effect modification into DAGs. I will post the pre-print link here when done. Of note, a drug may work through a receptor to mediate a target effect e.g. GIP analogue through the GIPR, but the drug does not “cause“ the receptor as it exists whether or not the drug is present. Even though the drug doesn’t cause the receptor, the receptor is unambiguously the mechanism of action, and thus a mediator of its effect.
We finally wrote the paper which we titled Effect Heterogeneity Due to Pathway Specific Treatment Effects and what we finally concluded surprised us as we did not expect we would end up here. The DAGs I previously suggested are clearly not the right ones and neither are those in previous papers like the one that started this thread.
Look forward to comments and feel free to be as radical as necessary.
Addendum
So now our preprint provides an answer to this question. The question has correctly identified that subgroup analyses in RCTs can produce biased interaction estimates, but its source has been misdiagnosed as unmeasured confounding. The problem is more fundamental and does not require any unobserved variable. In an RCT, randomization ensures treated and untreated groups are comparable before treatment, but if S is a post-randomization variable that is both associated with T and independently prognostic for the outcome, then conditioning on it immediately breaks that protection. The T×S interaction term is estimated within cells defined by the joint realization of treatment and say a subgroup biomarker response, and within each cell treated and untreated patients have arrived by entirely different causal routes → treated patients via treatment-induced biological response, untreated patients via background biology. This makes the within-cell comparison structurally incoherent regardless of whether unmeasured confounders exist, because the collider is opened by the analytic operation itself rather than by any missing variable.
The collider structure Y ← T → C ← S → Y exists in an RCT just as it does in an observational study. Randomization eliminates backdoor confounding paths into T but it does not and cannot eliminate the distortion introduced by conditioning on a post-randomization collider. The subgroup defined by T and S jointly is not a clean biological subgroup in an RCT any more than it is in observational data. This is why the problem is structural and irremovable, it is a consequence of the analytic operation of conditioning on S, not of how T was assigned. Note that C is not a separate variable in the world, it is the analytic act of categorizing patients by their joint T and S status and comparing outcomes across those categories. Every patient who enters the analysis has been selected into a cell by the joint realization of their treatment and their response to such treatment. The distortion is not a general property of all post-treatment variables. It is specific to post-treatment variables that are simultaneously treatment-associated and outcome-prognostic. These are precisely the variables that are most clinically interesting and most likely to be analyzed as mediators or effect modifiers. The collider is not something imposed on the data from outside. It is created by the analytic operation itself and the moment we condition on both T and S to define comparison groups, this collider opens. This means the selection distortion is not a feature of bad data or unmeasured confounding. It is an inevitable consequence of the analytic strategy, which is precisely why it is irremovable by any subsequent adjustment.
The consequence, demonstrated empirically in our paper, is that the interaction term does not estimate a stable biological quantity and it can attenuate, disappear, or reverse across trials drawn from the same population with identical underlying biology, not because the biology changed but because outcome events happened to fall differently across the four T×S cells in each sample.
The solution, which we discuss, is not to refine the interaction model but to replace it: define treatment as a three-level variable capturing pathway-specific (not path-specific which is a variant of mediation analysis) engagement, estimate each pathway effect against the untreated reference without S in the outcome model, and interpret the difference between pathway-specific effects as the direct empirical consequence of differential mechanism engagement rather than as a statistical interaction.
Excellent. I can follow this logic and believe it to be sound and intuitive. To clarify, this doesn’t apply to subgroups that are defined by pre-treatment variables? These can still be influenced by U as in the original DAGs at the start of this thread?
Yes, that is correct, the subgroup has to be based on a post-treatment variable or its proxy (the proxy could be a pre-treatment variable but must impact a post -treatment variable). For this scheme to hold, the subgroup must be based on a conditional mediator. The reason why we call the conditional mediator an effect modifier is detailed in the preprint.
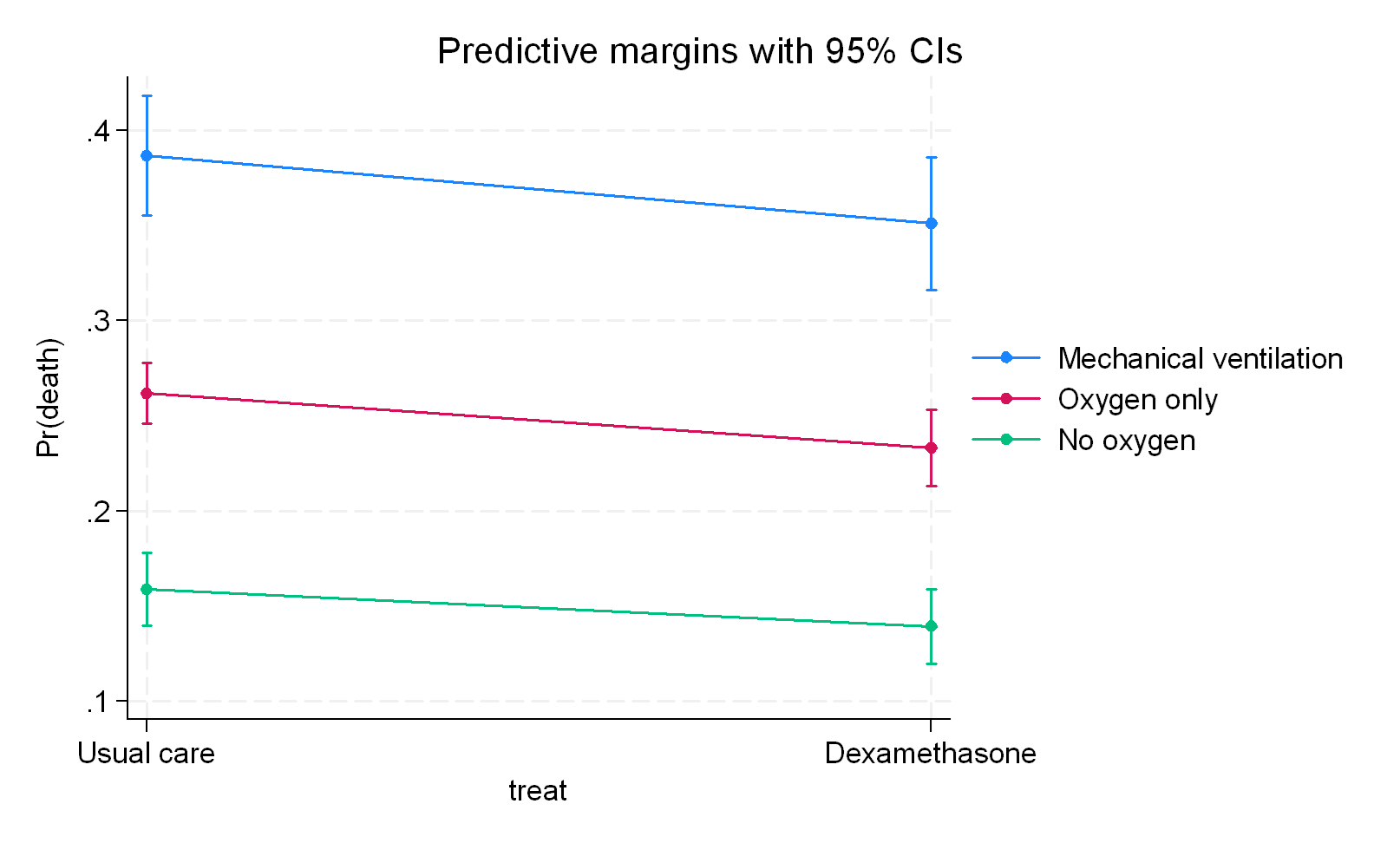
Where this would not apply are prognostic or clearly pre-treatment variables. For example in the RECOVERY trial of dexamethasone for survival in COVID there were subgroups based on mechanical ventilation (MV), on oxygen only (O2) and not on oxygen (no-O2). Here these are clearly prognostic and pre-treatment subgroups so applying this would fail completely. That said, I personally don’t believe in stratification for subgroup effects (meaning adding a product term to regression) and believe that adding the subgroup as a covariate suffices because these represent risk magnification if pre-treatment rather than effect modification. Of course, I may have missed something, so please correct me if you have any other thoughts about this
Because both are post intervention variables and therefore the only difference between mediators and effect modifiers are that with a mediator we have the structure for example:
Statin-> LDL reduction → CVD event
With the effect modifier we have the same structure with the following condition added on:
Receptor status ----> LDL reduction
So I believe that we can gate all mediators and once gated it is not induced by the treatment but remains associated with it through the post-treatment effect it induces. The gating variable is thus a conditional mediator and as I said above, every patient who enters the conditional analysis has now been selected into a cell (Receptor status x statin treatment cells) by the joint realization of their treatment and their response to such treatment, even if such response is replaced by the gating variable (the receptor status) and the response to treatment is not visible as that is a proxy for the gate. The distortion is not a general property of all post-treatment variables. It is specific to post-treatment variables that are simultaneously treatment-associated and outcome-prognostic. This selection bias seems to me to be irrecoverable if the receptor status enters the analysis, even though it is pre-treatment.
Of course if you have other thoughts about this, I would be keen to hear this.
If the variable is pre-randomization and solely prognostic for the outcome, then adjust in the analysis. All that stratification aka subgrouping (or adding a product term in regression which is the equivalent of stratification) achieves is overfitting and nothing else useful can be gained from this procedure. This is a form of effect heterogeneity called risk magnification.
To be an effect modifier, the variable must be associated with the intervention in a post-randomization sense as well as prognostic for the outcome, otherwise how would it modify the effect of the intervention on the outcome?
I have ceased to understand what we are discussing in this thread. I thought we were discussing how DAGs could assist in helping us examine whether there is potentially a differential effect of treatment within subgroup and whether there are potential confounders U that would also need adjusting for to identify this. You have written a paper that shows problems with this framework if the subgroup is defined by a pos-randomization variable but most of us were considering pre-randomization subgroups. I understand this comment to mean that you do not believe these DAGs or the resulting adjustment sets are beneficial to find treatment effect heterogeneity? This seems to go against the thesis of this thread. Care to elaborate?
As a clinician/researcher for 40 yrs one of the most important thing I have learned is to distinguish real biological data-generating processes (BDGPs) from synthetic data-generating processes (SDGPs). An SDGP is a gate-generated analytical construct that does not correspond to a single coherent biological causal system.
Your paper is provocative and deeply insightful because it appears interpretable within the SDGP framework as describing the creation of a post-treatment SDGP. Traditional cause-agnostic syndrome RCTs create SDGPs upstream through consensus enrollment gates (e.g., sepsis, ARDS), whereas under your framework treatment-responsive subgrouping appears to create SDGPs downstream by conditioning on treatment-associated pathway states or biomarker responses.
In both cases, the analytical population is generated by conditioning on a gate rather than by identifying a coherent biological causal system. The resulting estimands become structurally unstable, composition-dependent quantities that may reverse or vary despite unchanged underlying biology.
Thus syndrome disease-mixing and post-treatment response grouping may represent parallel manifestations of synthetic gate conditioning occurring at different temporal locations within the causal structure.
An important implication is that pre-specification of a subgroup may not rescue the analysis if the subgroup itself constitutes a SDGP. Pre-specification does not eliminate structural instability arising from synthetic conditioning.
Thank you both for the comments. I agree it is confusing so let me try to make things clearer using the RECOVERY data.
a) If I assume that ventilation status is a pre-randomization prognostic variable then I add it to regression as an adjustment and get a benefit across all three groups:
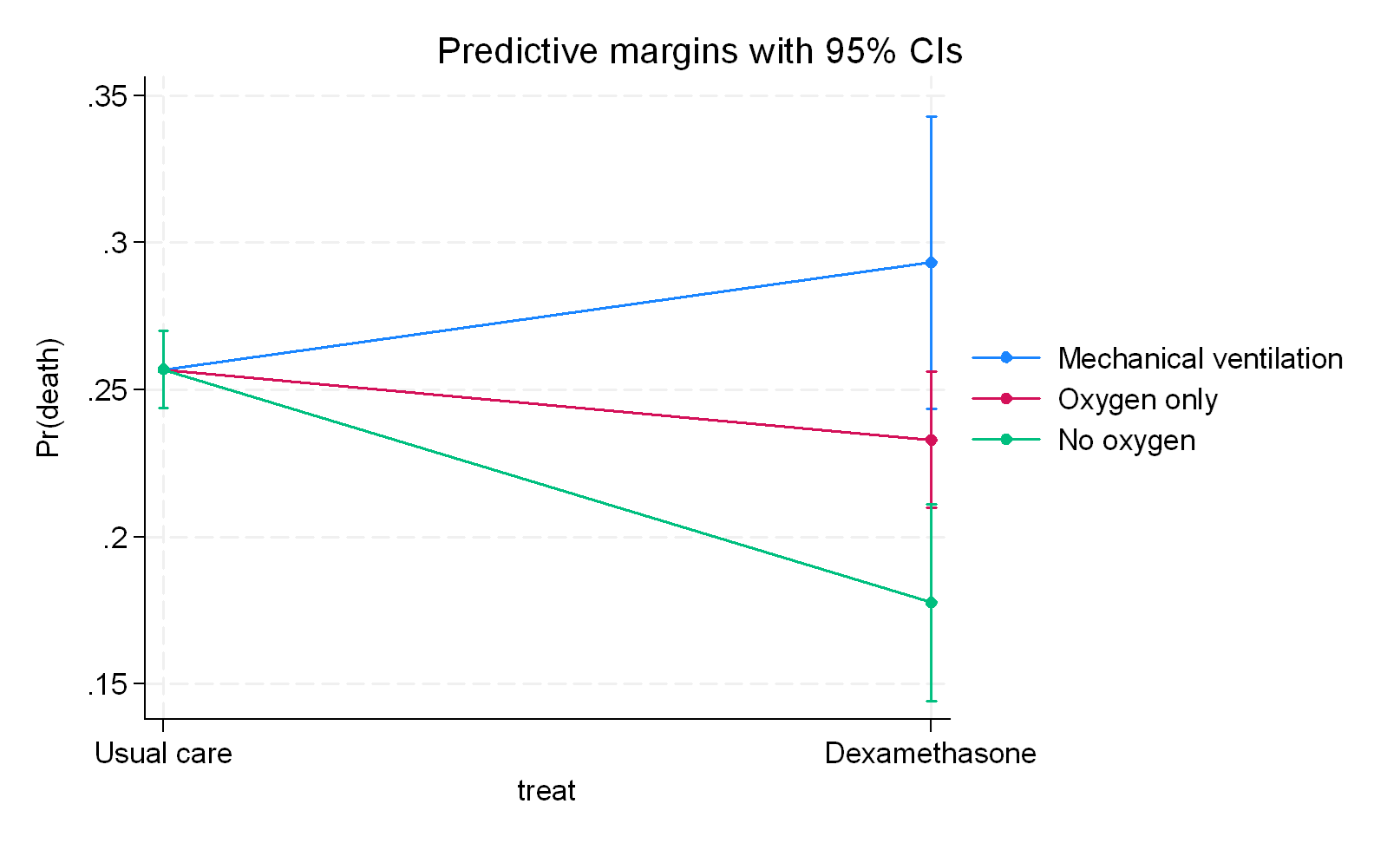
c) If I consider the ventilation status as a pathway variable (newly introduced in the preprint) then we get the following that means that the pathways differ from the no-intervention in different ways as follows and also assumes no pathway without intervention:
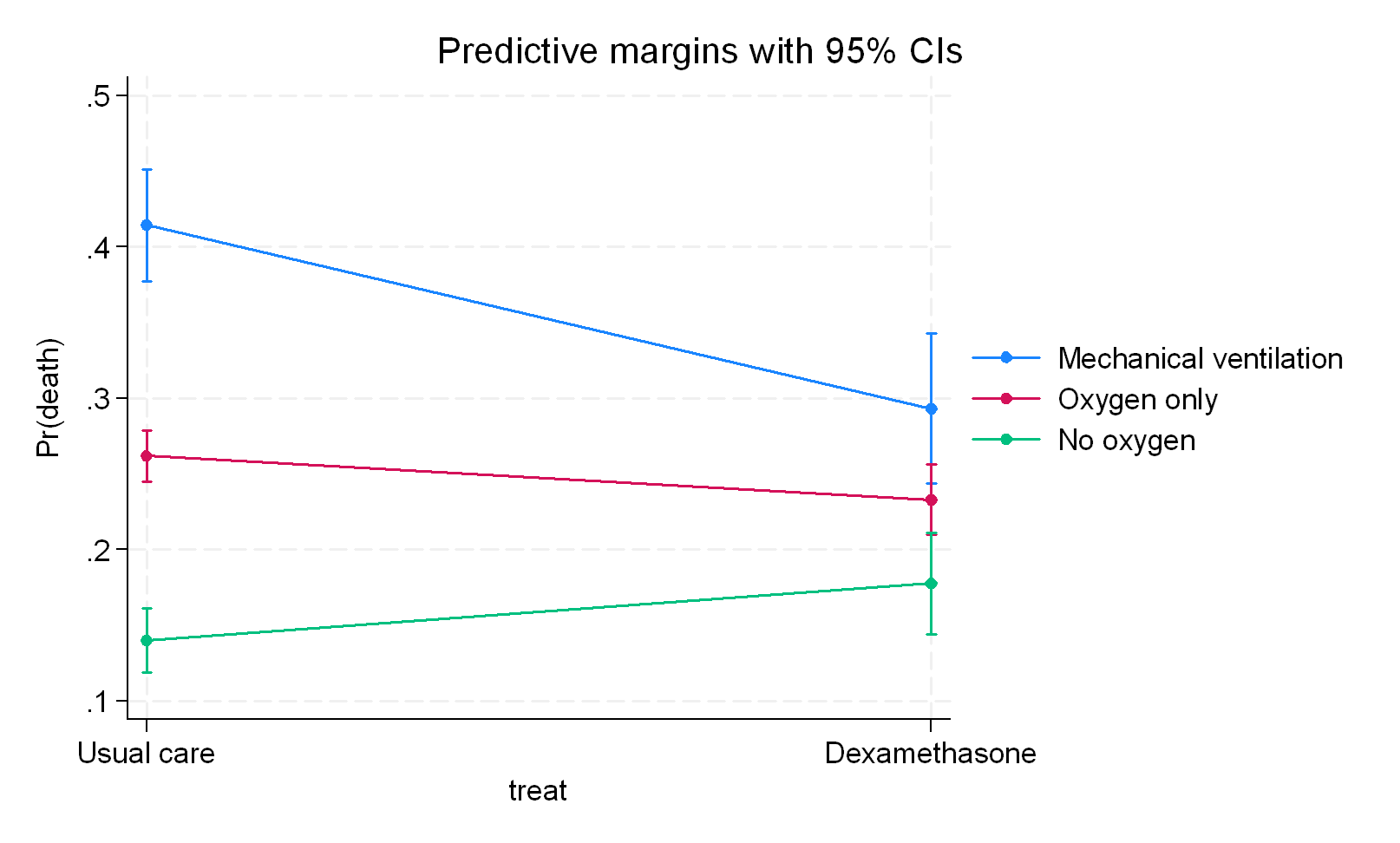
What we infer clearly depends on where we position the third variable structurally meaning that we need to be certain what we are dealing with here. I would go for a) with RECOVERY but the authors went with b) and all I am saying is that b) just represents overfitting because there is no clear indication in my mind that ventilation status represents a variable associated with dexamethasone in a post-randomization sense nor is there any indication that c) is correct which is that it serves as a pathway only for the intervention. Even had the modifier criteria been accepted, a further argument I make is that a subgroup or product term for ventilation status would be wrong because its entry into the outcome model would lead to collider (selection) bias.
The RECOVERY data nicely summarizes the preprint so you can now decide if this sits with you or not.
I don’t follow. Do you mean b to be a subgroup variable acting as an effect modifier or
a subgroup variable being post-randomization or treatment-associated?
The variable does not change - what we do with it does. In b) we treat it as an effect modifier and these results are observed. Now we have to decide if this analysis is valid and that decision hinges on what the variable is concluded to be and if the specific analysis is accepted as the way to analyze such a variable or not.
In b) we assumed effect modifier and stratified - I disagree with both decisions