I have read the paper by Pearl and @scott again following further recent discussion on Twitter by @stephen and @f2harrell. In their introduction on page 2 (see https://ftp.cs.ucla.edu/pub/stat_ser/r513.pdf ) Pearl and Scott suggest in their ‘Model2’ that a drug can both kill and save at the same time (i.e. “Model-2 – The drug saves 10% of the population and kills another 10%”) However, this could not work via the same causal mechanism, which either increases the probability of death (by killing some people) or reduces the probability of death (by saving some people). In order to do this via the same mechanism, the drug would have to create both its outcome and its counterfactual outcome at the same time.
We must therefore postulate two different mechanisms (e.g. a drug saving people through the mechanisms of killing cancer cells but killing people through the mechanisms of destroying their bone marrow). We can envisage this happening by failing to control the dose of a drug properly so that the bone marrow is wiped out by too high a dose leading to death from overwhelming infection before the drug can kill the cancer cells. Some people might avoid this fate by being physically large so that the amount of drug distributed in their system is appropriate. Men would therefore fare better than women and large people of either gender better than small people.
In a randomised controlled trial, there would be careful supervision, especially of the drug dosage where its therapeutic window was very narrow. (A therapeutic window means that too low a dose does not work, too high a dose kills but a correct dose saves lives.) This means that in a RCT the correct dose would be calculated so that the only effect seen was the killing of cancer cells, thus reducing the proportion dying compared to no drug treatment. In such a carefully conducted RCT there would be no deaths from an adverse drug effect. The result of such a RCT is shown on the left hand side of Table 1, which is taken from the data in Pearl and Scott’s paper. There is a reduction of 28% in the proportion dying (49%-21% =28%) and an increase in 28% of people surviving (79%-51%=28%).
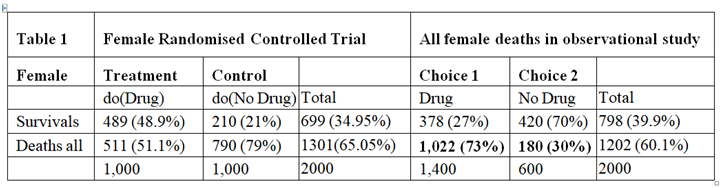
The right hand side of Table 1 describes the outcome of the observation study where 1400/2000 = 70% chose to take the drug and 600/2000 = 30% chose not to take it. In this observation study, there is an INCREASE in the proportion dying in those who take the drug by 43% (73%-30%=43%) and a corresponding reduction of 43% in people surviving. The observation study was done on people who chose for themselves to take the drug by self-medication without medical advice and supervision. We can imagine that they were therefore at risk of not taking the correct dose according to their body weight. If the drug had been taken correctly, then the proportion of 30% dying with no drug should have been lowered by 28% to 30-28=2%. The result of the observational study would then have been that shown in Table 2, where the deaths would have been due to disease alone and much reduced to 2% and the proportion surviving much increased to 98%.
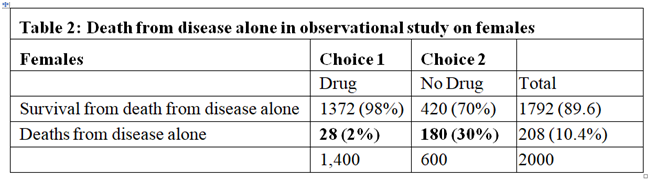
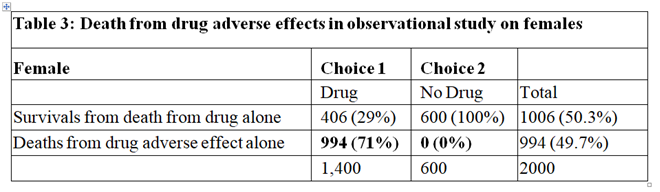
In the observational study, of those who took the drug 73% died. From Table 2 only 2% were expected to die of the disease when taking the drug, this suggesting that 71% who died on the drug were due to its adverse effects. Therefore 71% of those on the drug were harmed by being killed by it as shown in Table 3. Clearly of those who did not take the drug no one died from the adverse effect of the drug, as also shown in Table 3. By adding the cells in bold in Tables 2 and 3 we get the cells in bold in Table 1.
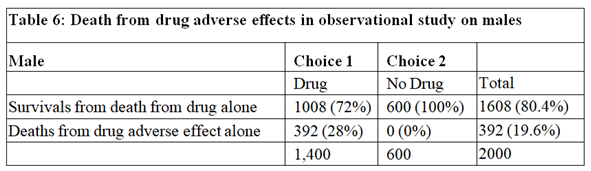
The data for males are shown in Tables 4, 5 and 6. Tables 2 and 5 are identical because if there had been no deaths from adverse effects, the outcome of the observational study based on similar RCTs results would have been the same in males and females.
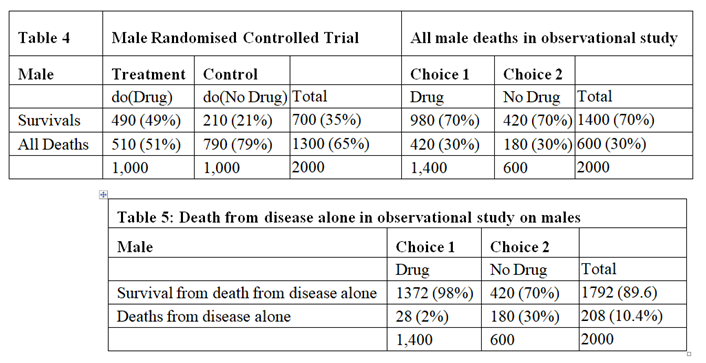
The deaths from adverse effects were much lower in males (it was 28%) compared to females (it was 71% in females), perhaps explained by the higher body mass of males, giving greater protection for high tissue levels of the drug despite taking too high a dose.
Conclusion
By postulating different causal mechanisms for harm and benefit, the proportion of females harmed by the drug was 71% and the proportion of males harmed by the drug was 28%. These rates are very different to the results of Pearl and Scott who reasoned that the proportion of females harmed was 0% and the proportion of males harmed was 21% (see page 10 on https://ftp.cs.ucla.edu/pub/stat_ser/r513.pdf ). However, these results agree with Pearl’s and Scott’s view that observational studies subsequent to RCTs can provide useful information especially on the adverse effects of treatment in the community perhaps due to errors in drug administration and especially when adverse effects are rare and unlikely to show up in RCTs on comparatively small numbers of subjects. Such observational studies are of course well established practice.