I’m analyzing TCGA survival data to understand the prognostic importance of biomarkers from genes G1 and G2. Two different markers are measured for each gene: copy number (cnv) and expression (rna). I’ve fitted a pre-specified model which includes the genetic markers and other prognostic factors available from the data:
test.model <- fit.mult.impute(Surv(OS.time, OS) ~ rcs(age, 3) + gender + tobacco_smoking_history +
radiation_therapy + G1.cnv + G2.cnv + rcs(G2.rna, 3) * rcs(G1.rna, 3),
cph, imp, data = patients)
Checking the ANOVA for the model yields:
Wald Statistics Response: Surv(OS.time, OS)
Factor Chi-Square d.f. P
age 2.89 2 0.2359
Nonlinear 2.71 1 0.0999
gender 3.73 1 0.0533
tobacco_smoking_history 2.38 4 0.6670
radiation_therapy 1.42 1 0.2330
G1.cnv 0.70 4 0.9519
G2.cnv 0.54 4 0.9700
G2.rna (Factor+Higher Order Factors) 14.06 6 0.0290
All Interactions 13.89 4 0.0077
Nonlinear (Factor+Higher Order Factors) 6.42 3 0.0930
G1.rna (Factor+Higher Order Factors) 14.36 6 0.0259
All Interactions 13.89 4 0.0077
Nonlinear (Factor+Higher Order Factors) 2.34 3 0.5040
G2.rna * G1.rna (Factor+Higher Order Factors) 13.89 4 0.0077
Nonlinear 6.91 3 0.0749
Nonlinear Interaction : f(A,B) vs. AB 6.91 3 0.0749
f(A,B) vs. Af(B) + Bg(A) 0.03 1 0.8723
Nonlinear Interaction in G2.rna vs. Af(B) 5.80 2 0.0550
Nonlinear Interaction in G1.rna vs. Bg(A) 0.85 2 0.6534
TOTAL NONLINEAR 9.65 6 0.1402
TOTAL NONLINEAR + INTERACTION 15.57 7 0.0294
TOTAL 27.56 24 0.2792
The small p-values for G1.rna, G2.rna, and the interaction(0.0259, 0.0290, 0.0077 respectively) suggest to me that the expression markers for each gene may have prognostic value, but I’m concerned that the model’s overall p-value is rather large (0.2792).
I’ve further assessed the importance of G1 and G2 expression with the likelihood ratio test and
the adequacy index:
subset.model <- fit.mult.impute(Surv(OS.time, OS) ~ rcs(G2.rna, 3) * rcs(G1.rna, 3),
cph, imp, data = patients)
lrtest(subset.model, full.model)
A <- 1 - subset.model$stats['Model L.R.'] / full.model$stats['Model L.R.']
lrtest p-value = .1 and A = .42, providing some (although weaker) evidence that G1 and G2 expression may be prognostic.
How should I interpret these contrary results? Is it possible to reconcile the overall model’s large p-value with the individual variables smaller p-values, or must I conclude more data is needed to make any conclusions? In general, if the overall model’s p-value is large, should any individual p-values be ignored?
Additionally, I’m trying to visualize the relationship between G1.rna and G2.rna. When I visualize all the predictors the plots seem rather flat:
ggplot(Predict(test.model))
Visualizing only G1.rna with certain values of G2.rna is much less flat:
ggplot(Predict(test.model, G1.rna, G2.rna = c(0, 3, 6, 9)))
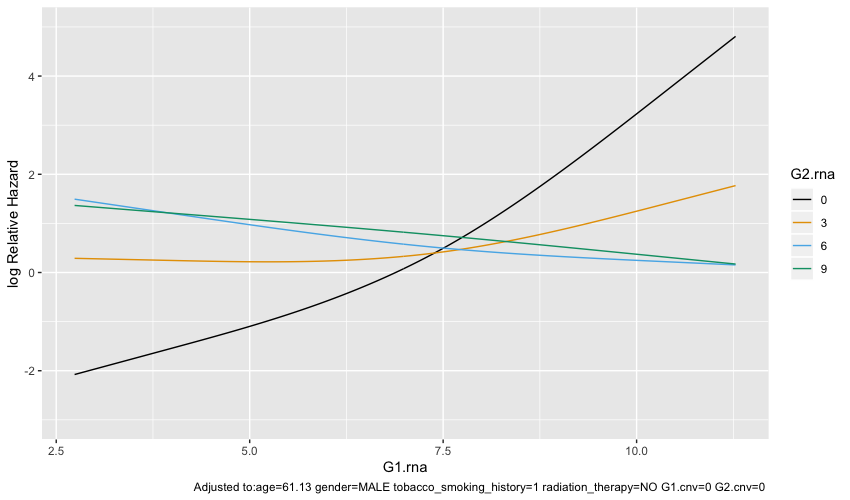
I see that the the second plot says adjusted to several factors while the first plot doesn’t. Are the curves from ggplot(Predict(test.model)) averaged across all conditions while ggplot(Predict(test.model, G1.rna, G2.rna = c(0, 3, 6, 9))) shows the curve only for specific condition? If so, which plot should I be showing?