Hi,
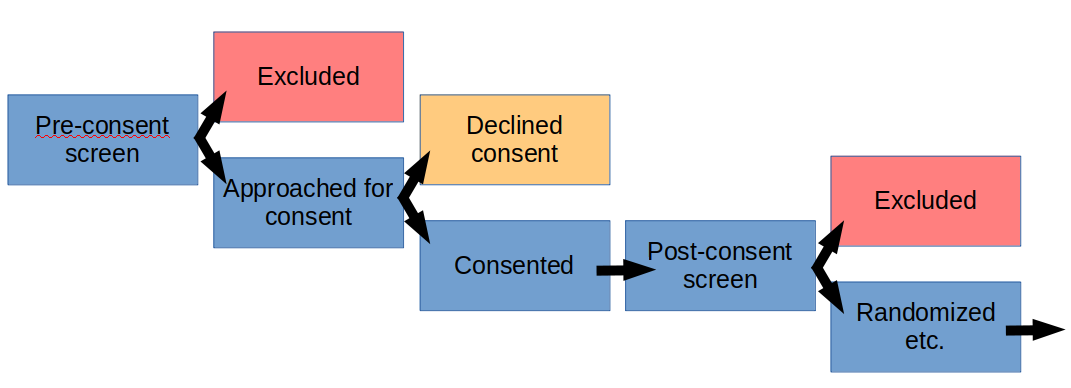
Typically, as part of a study, there will be pre-defined patient recruitment and screening procedures that have been approved by an IRB and possibly by other regulatory bodies, if relevant. That includes the manner of patient contact, which may be via telephone, in-person, electronic, or other means.
The details may vary from institution to institution, but generally, you can create and maintain a screening log of patients that have been screened, and are not eligible and/or declined to participate. The IRB would typically provide a waiver to support this activity, to cover the collection of pre-defined data, that may or may not contain PHI, depending upon functional requirements.
If the patient declines participation, typically, any PHI that may have been collected during screening would be destroyed, unless there are overarching reasons to maintain it and that has also been approved by the IRB.
As always, the data that can be collected and maintained in the screening log should meet the minimum necessary standard to fulfill the needs of the approved study. Since this will typically take place prior to the signing of an informed consent for the study, you would want to minimize the amount of data that you record.
If you need to conduct study specific diagnostic testing to confirm eligibility for the study, then the informed consent would need to be signed and that document would typically have some IRB approved language regarding screening procedures and authorization to conduct the testing and to collect specific information.
Gender, race and age would not be PHI, unless the age is over 89, in which case, you need to create a category of “age >89” and not track the actual age in years for those patients. Thus, you would not need patient approval to collect and retain those.
The reason for declining to participate would also not be PHI, and you might have a pre-defined set of reasons that are available for consistency in collection, as opposed to free text.
There are published studies regarding patient recruitment, selection bias, why patients decline to participate, etc. So, IRBs do approve the collection of such data when there are valid reasons to do so.