The purpose of this topic is to start a discussion about the methods used in the Mayo Clinic paper and how the paper has been interpreted and misinterpreted by the FDA. It is hoped that others will write more in-depth evaluations of the paper’s methods. Here are some initial thoughts.
The authors provided a list of reasons why it was not possible to randomize patients to CP from the start. These reasons are singularly unconvincing.
There is no control group in the paper so the authors and FDA relied on evidence for an association between earlier transfusion and waiting time for transfusion <= 3 days having a lower 7d or 30d mortality than transfusion done at 4+ days, and a putative “dose-response” relationship between IgG antibody levels of donor plasma and mortality.
The study design assumes there are no deaths before 4 days, and I can’t find data on frequencies of death by day. I am also uncertain about the definition of “time zero”.
By categorizing transfusion time and IgG the authors failed to provide the needed information with enough resolution, and left the analysis open to gaming as discussed by Howard Wainer who showed that from the same set of data cutpoints can be found that yield a positive association and different cutpoints can be found that demonstrate a negative association. It is very unlikely that the authors engaged in such gaming but there was no related documentation in the paper.
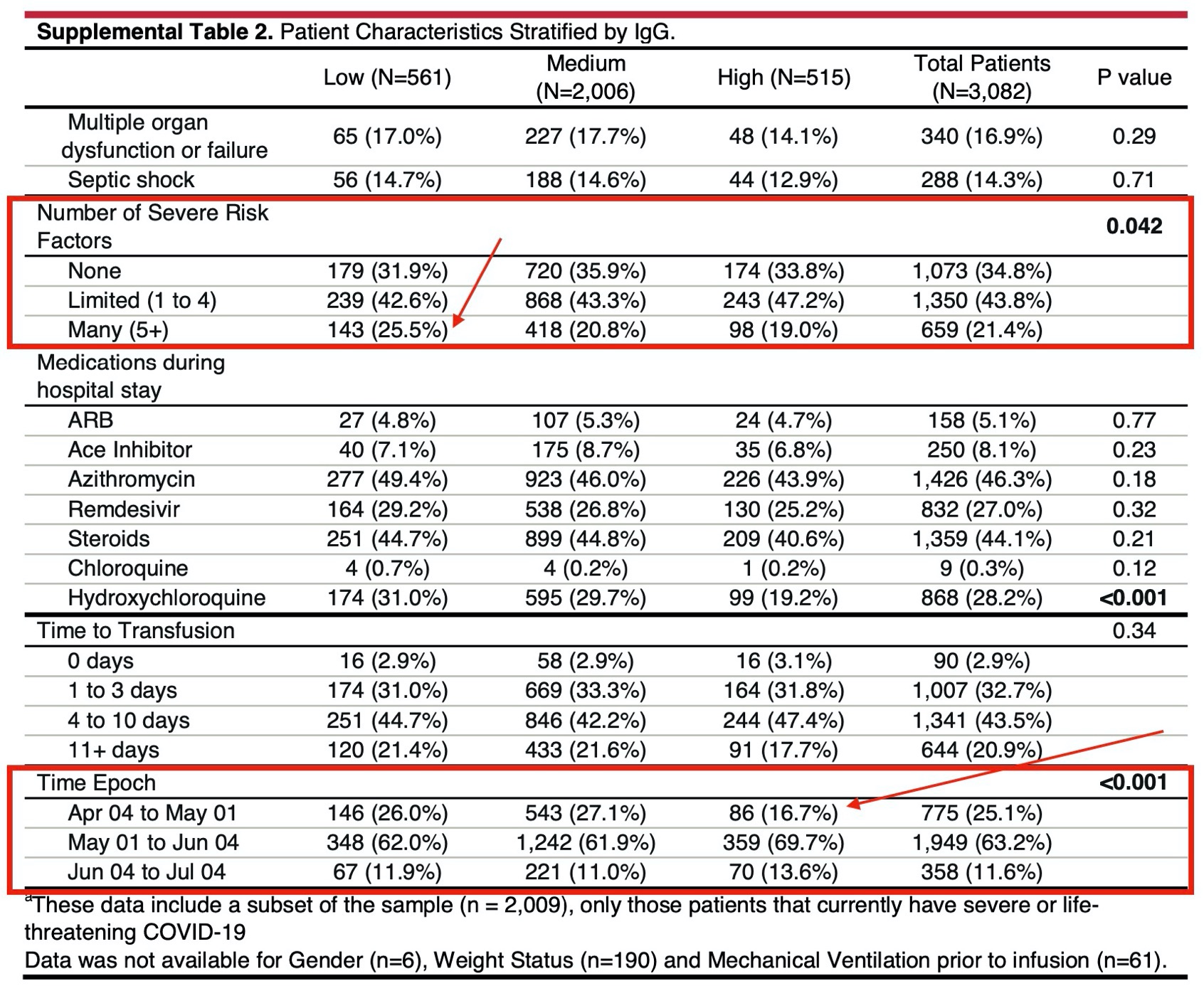
The “low”, “medium”, and “high” IgG level intervals used in the paper involve intervals that are too wide resulting in much heterogeneity within the intervals.
IgG levels were available in < 0.1 of the patients and the authors did not do a propensity analysis to show predictors of having IgG levels determined as a function of patient baseline characteristics.
The authors lost a major opportunity to provide the reader and FDA with continuous joint analysis of IgG levels and day of transfusion using e.g. logistic regression with flexible nonlinear effects of IgG and transfusion day.
The time-dependent nature of the treatment (transfusion) was not explicitly used in the analysis. Transfusion is an “internal time-dependent covariate” which presents special analytical problems. The authors seemed to analyze the data as if transfusion is a baseline covariate.
As @Biomaven suggested below, the analysis needs to adjust for a secular trend. This can be done by included in a model a restricted cubic spline in days from 2020-01-01 with 7 knots to allow for changing background therapy.
Likewise, in doing a propensity analysis of IgG being measured, calendar time should be flexibly included in the binary logistic model.
There is confusion about relative vs. absolute treatment effectiveness estimates. When referring to a 37% reduction in mortality, I recommend that the two absolute risk estimates be emphasized and the 37% be greatly de-emphasized.
An FDA tweet made the patently false claim that CP benefits 0.35 of patients. As @Stephen has written many times, a parallel-group study simply cannot do what a 6-period randomized crossover study can do: provide an estimate of the proportion of patients who benefit from a treatment. Parallel-group data cannot distinguish all patients getting a small benefit from 0.35 of patients getting full benefit and the remaining 0.65 getting no benefit. There is absolutely no way to get the 0.35 figure from the Mayo Clinic paper.
My Vanderbilt colleague Jill Pulley provided the following question and an interesting way to think about how a study without a control group may mislead:
The part I don’t understand even from the Mayo paper is how they could say it is consistent with efficacy when it could also be consistent with the low titer units being unsafe. The Mayo study measured donor antibody levels in sera using the Ortho-Clinical Diagnostics VITROS Anti-SARS-CoV-2 IgG chemiluminescent immunoassay. This is qualitative assay based on a recombinant form of the SARS-CoV-2 spike subunit 1 protein. Results of this assay are based on the sample signal-to-cut-off (S/Co) ratio, with values <1.0 and ≥1.00 corresponding to negative and positive results . However, they created a “semi-quantitative” interpretation of this assay and established relative, low, medium and high binding antibody levels by setting thresholds for low and high based on the ~20th and ~80th percentiles of the distribution for the S/Co ratios, respectively. To us, a key issue to consider in this approach is that if their ‘low’ category included sub-therapeutic doses with non-neutralizing concentrations, rather than being removed from the pool of available plasma (as in our trial), these can theoretically trigger antibody-dependent enhancement (ADE) which can facilitate virus uptake and subsequent worsening of symptoms. For this reason, doesn’t it seem the group receiving ‘low’ titer plasma could have actually been made WORSE, causing the ‘high’ group to look relatively efficacious and fostering an erroneous interpretation of the results???
A dose-response study without a control group is analogous to having an interaction in a regression model without having the main effect. The interaction analysis may show the slope of the dose in the right direction, but the lack of main effects causes the intercepts to be incorrect, so that you don’t know where to vertically position the line.
The redaction of FDA personnel names in the FDA EUA memo is troubling:
The authors’ narrative conceit is that benefit day 1-3 compared to day 4 shows the treatment works. This alone is highly debatable. But even accepting it, they do none of the things you’d want to do to show that the difference is meaningful. There’s no table comparing early and late treated patients, no multivariable analysis, No fixed effects model, no adjusting for time, nothing except a single stratified table. Especially Since we’re finding most of these really matter to outcomes, these are very basic limitations.
Agree that showing treating earlier better than later is not equivalent to CP works compare to standard care. In this regard, I think a comparison with historical data (using matching or other techniques) may be helpful.
Also, I noticed that Dr Hahn, at the pressor, compared the statistics reported in this paper to the survival analysis usually use in oncology studies. I don’t think this is very accurate either. While it is true that mortality rates are often reported in survival analyses, the primary focus of survival analyses is the survival time rather than survival rates.
They adjusted for “disease severity and demographic features” but apparently not for epoch. Given the clear change in patient population and treatments over time (more remedesivir, more steroids, less HCQ, quicker use of CP), should they not present the analysis by epoch separately?
In particular, as the patient population got less critical, they also speeded up the time to use CP, so there is clear confounding on this aspect.
Great point. I suggest that they adjust for a restricted cubic spline in calendar time (days from 2020-01-01) with 7 knots to allow for a lot of flexibility around the times that concomitant meds changed.
Without getting into the interpretation of the data by others including colleagues at FDA, my main two worries from a scientific perspective are:
Residual confounding by indication: many methods exist to minimise or at least test for the presence of unobserved confounders. None seems to have been used here
Potential for immortal time bias and similar issues with ‘time zero’ definition. The definition of the ‘time zero’ (aka ‘index date’) for both arms is insufficiently detailed in the preprint. I hope this can be clarified in the full manuscript.
@f2harrell: To your question Frank, about the source of the 0.35 figure, Trump quoted that in his presser, as did Azar, and there is an article on StatNews that includes some questions as to where that figure comes from:
There are also some comments in the above article regarding the redaction of the FDA staff names, and an implication that this may have been done because they do not support the findings as presented.
There is also an editorial on StatNews by Art Caplan, raising the implications of the EUA:
Someone said that someone said that 35% of patients will be benefited. I can’t nail down that quote. But what is nailed down is that the commissioner wrongly said multiple times that of every 100 patients 37 will be saved by CP. A gross misunderstanding of a relative risk of 0.63.
Frank, there are some late updates tonight to the Washington Post article that you linked to above and in a NY Times article on the same subject, with both now including relevant tweets and new comments from Hahn:
The analysis is done with days after diagnosis (I presume the positive PCR this is) as the primary covariate. This means that the diagnosis can thus be made from >10 days preceding hospital admission (if the test was done by a GP of testing venue) up to many days after hospital admission.
Important bias may happen when patients who get tested easily / earlier on in the disease course while still outside the hospital also can get hospitalized easier (e.g. good health insurance). These patients will be overrepresented in the “treatment within 3 days after diagnosis” group. I do not see how this was (and can) be accounted for.
The study should therefore analyse the treatment effect in function of symptom duration at time of plasma transfusion and how this relates to a possible therapeutic effect of plasma. Hope this will be possible
entirely agree with this point @bjarijn and very much in line with my point re ‘time zero’ above. Getting time zero/index date wrong or ill specified is the single most common origin of bias and trouble in pharmacoepi.
As suggested by Miguel Hernan, James Robins and others, if you cannot quite visualise the trial you are emulating you cannot answer a causal question…
The availability of high titer plasma and earlier treatment with plasma increased as months passed, as did treatment with known effective treatments (remdesivir and dexamethasone). Ineffective treatments that may have caused harm, such as hydroxychloroquine were used less frequently in later months. Finally, and perhaps most importantly, the experience of intensivists in treating COVID-19, particularly respiratory failure, improved over time. Less invasive and dangerous forms of respiratory support were increasingly employed. Thus the failure to do a simple multivariate analysis or Cox regression including these factors, or at least date of diagnosis means we know very little about what factors were associated with survival. One last thought is that some plasma was ABO “compatible” which has been associated with increases in sepsis and acute respiratory failure in observational studies, compared with ABO identical plasma. This factor was not addressed at all and needs to be analyzed at some point. The field of transfusion medicine has failed to accept both randomized trial and observational data demonstrating a relationship between the ABO identicality of a transfusion and important clinical outcomes, including bleeding, organ failure and mortality.
I’ve posted a comment on this preprint at medRxiv, referencing some of the points raised here, with some additional points that haven’t been raised here yet.
It’s often easy to pick apart studies (especially observational studies). Does this one have any value? Is there anything we could learn from it to inform future research?
 My Vanderbilt colleague Jill Pulley provided the following question and an interesting way to think about how a study without a control group may mislead:
My Vanderbilt colleague Jill Pulley provided the following question and an interesting way to think about how a study without a control group may mislead: