Hi!
I have longitudinal data of abdominal surgery patients at 4 timepoints (baseline, 1-month, 3-month, 6-month). The response is the EQ visual analogue scale (EQ VAS), which ranges from 0 to 100. The 3 groups for comparison are mild-to-moderate anemia, mild anemia and non-anemia. I have used the generalized least squares and followed the guide from the rms book/short course.
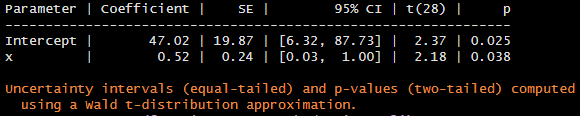
Here is the code that I have used.
a <- Gls(vas ~ anemia * month +
vas0 + rcs(age, 4) + sex + type + lap_open, data=df,
correlation=corCAR1(form = ~month | ic))
where, vas is the vas score at follow-up (i.e. 1-month, 3-month and 6-month), anemia is the 3-level severity of anemia (i.e. mild-to-moderate anemia, mild anemia and non-anemia), month is integer value (i.e. 1, 3, 6) vas0 is the baseline vas score, age is age of patient at baseline, sex (female, male), type is either gastro, hepa or uro/gynae, lap_open (i.e. either surgery is laparoscopic or open) and ic is the unique patient identifiers. I assume linearity for month. In the book, it stated “Time is modeled as a restricted cubic spline with 3 knots, because there are only 3 unique interior values of week.” Here, what does it mean by interior values?
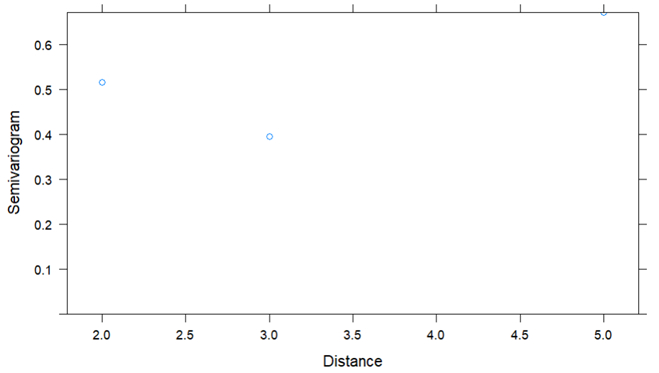
When I plot the variogram, it gives me only 2 points. How do I assess this?
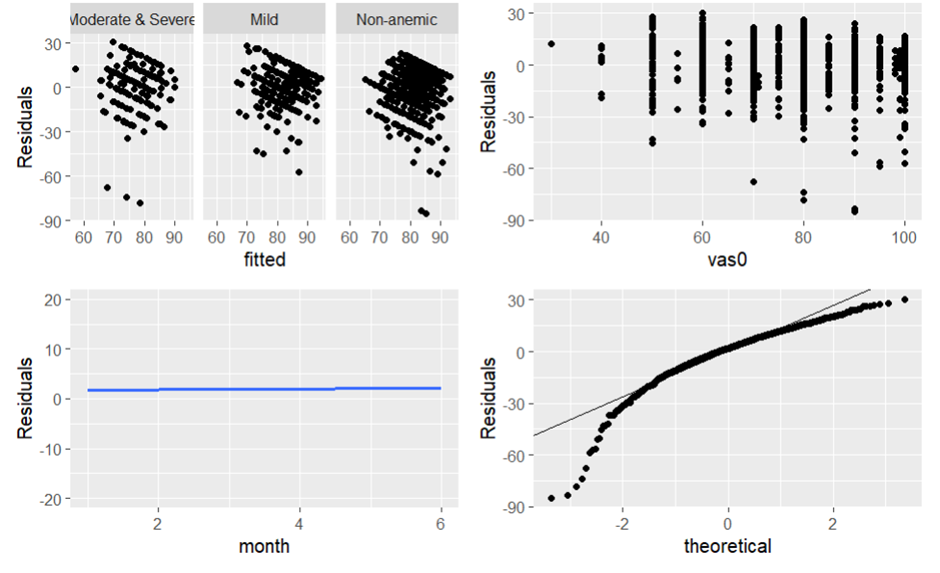
When I plot the variance and normality assumptions, the residuals do not look random and the qqplot does not seem to follow the theoretical normal distribution. Does this suggest that I abandon the GLS? What statistical method should I use here?
Besides EQ VAS, I would also like to look at the individual domains of the EQ-5D-3L (ordinal variable with 3 levels) and utility score (range from 0.854 for state 11121 to -0.769 for state 33333). Would it be useful to look at Semiparametric Ordinal Longitudinal Models?
Thank you!