I’m an early career researcher and long-time member of datamethods. I am an MD/PhD from a small country with a very small community of statistical and epidemiological expertise – which means that by necessity, I must do most of my statistical work myself. I have become enamoured with all aspects of clinical prediction modelling, from development to deployment. I and my team recently published a risk prediction model that we intend to extend and validate, and unlike many prediction models, are actively working on getting included into clinical guidelines for general use. Because there exists very little expertise in prediction modelling at my institution (and country) I receive very little critical feedback, neither positive or negative, and I am afraid that my work is not as rigorous as it could be. I wanted to see whether the datamethods community would consider reading my work and providing me with feedback, positive or negative. This could be considered community service, as for better or worse, my models are very likely to actually be used by clinicians and my research career will likely be spent producing them.
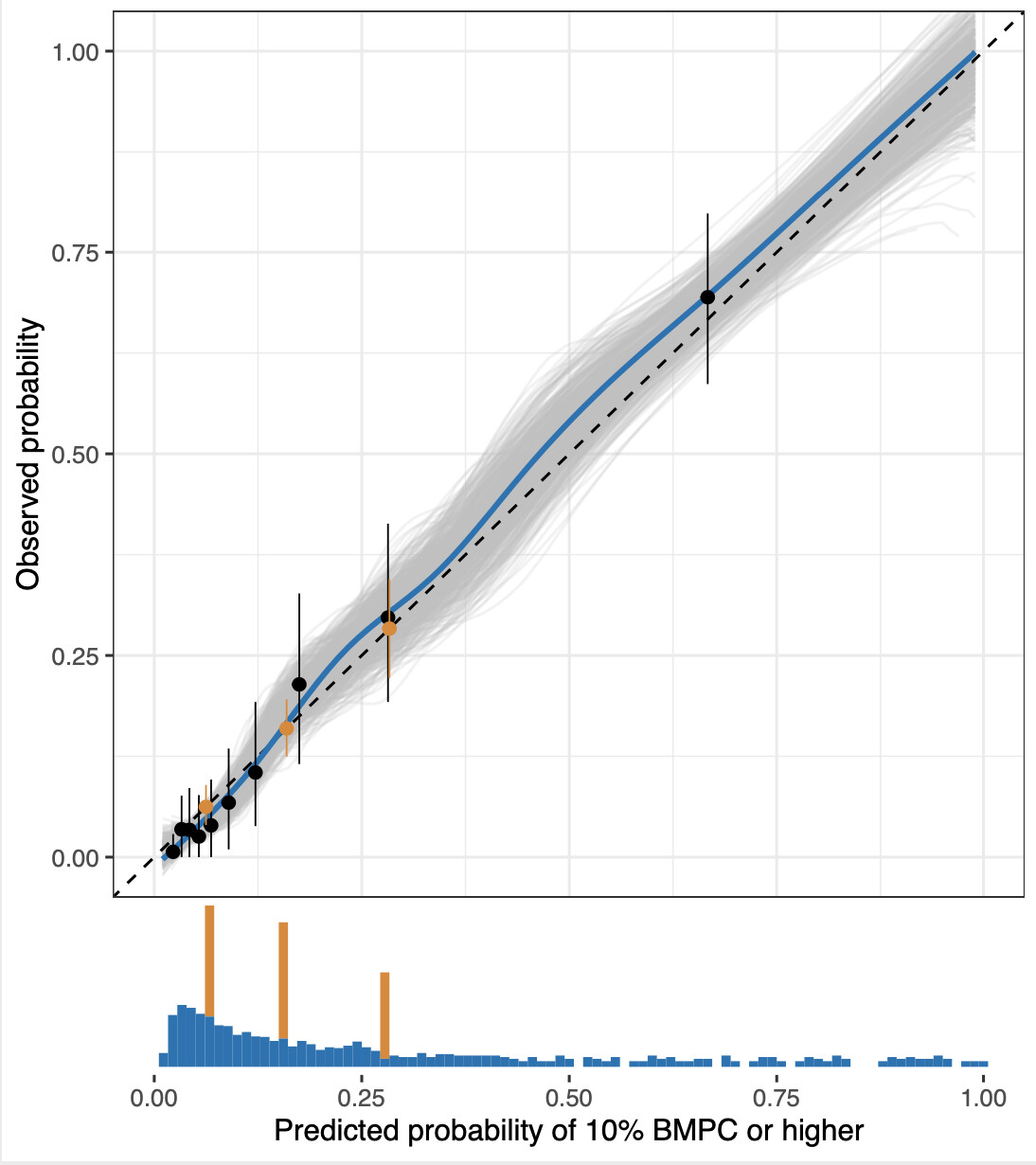
The model is described in Development of a Multivariable Model to Predict the Need for Bone Marrow Sampling in Persons With Monoclonal Gammopathy of Undetermined Significance: A Cohort Study Nested in a Clinical Trial https://www.acpjournals.org/doi/10.7326/M23-2540 . I will provide an attachment bellow that includes an early draft of the paper before the editorial process began for those who cannot access the publication through their institution (Edit: the editorial team vetoed this being shared). For a brief background: monoclonal gammopathy of undetermined significance (MGUS) is an asymptomatic precursor to multiple myeloma (MM) and other lymphoproliferative diseases. MGUS is extremely common, roughly 3-5% of adults 50 years of age and older. All individuals with MM are thought to go through MGUS, but the majority of persons with MGUS never progress to MM. MGUS can be diagnosed and much of the determination of the risk of progression can be completed with a simple blood test. However, the proportion of bone marrow plasma cells (BMPC) is also a significant risk factor. If BMPC are ≥10%, this defines an intermediate state known as smoldering multiple myeloma (SMM), which has a higher probability of progression to overt disease, such as MM, and clinical guidelines suggest much more intense monitoring for this group. Bone marrow sampling is an extremely safe procedure, but can be painful and is generally only performed in specialised centers. Our model predicts the probability of ≥10% BMPC (and therefore SMM by bone marrow criteria) based on commonly available parameters to inform the decision to refer persons with presumed MGUS for bone marrow sampling.
The original model was an ordinal logistic model with outcomes 0-4%, 5-9%, 10-14% and ≥15% bone marrow plasma cells and five variables (18 parameters): MGUS isotype (IgG, IgA, biclonal and light-chain, 3.d.f.), M protein concentration (modelled with a restricted cubic spline with four knots, 3.d.f.), free-light chain (FLC) ratio (modelled with a restricted cubic spline with four knots, 3.d.f.), and total IgG concentration (modelled with a restricted cubic spline with four knots, 3.d.f.), IgA concentration (modelled with a restricted cubic spline with four knots, 3.d.f.) and IgM concentration (modelled with a restricted cubic spline with four knots, 3.d.f.).
During the editorial process questions were raised with regards to
-
using an ordinal model compared to a logistic model (<10-14% compared to ≥10-14%). I justified this in
MGUS_prediction_ordinal_vs_logistic.html (2.0 MB) -
using restricted cubic splines for continuous parameters. We ultimately choose to include total IgG, IgA and IgM as linear variables, justified in
MGUS_prediction_rcs_vs_linear.html (3.9 MB) -
whether total IgG, IgA and IgM should even be included. We justified including these in MGUS_prediction_total_immunoglobulins.html (1.8 MB)
-
wether interaction terms between MGUS isotype and total IgG, IgA and IgM concentrations should be included. We argued against this in
MGUS_prediction_interaction_terms.html (3.0 MB)
MGUS_prediction_interaction_terms_linear.html (3.0 MB)
The clinical calculator is available here. I have requested permission from the editors of Annals of Internal Medicine to attach the manuscript here. Edit: This request was denied. Rmarkdown .html file of all the statistical code was to large to post onto datamethods so I uploaded it to OSF.
Any and all criticism, whether it be wording of the manuscript, methodological decisions, how the code was written or presented, or presentation of the clinical calculator is all greatly appreciated, no matter how pedantic or minimal.