One way to connect these would be to take for example a patient who has prostate cancer. X is the choice between a treatment or control (control here would be surveillance, i.e., no treatment). Y is the outcome of interest, which here can be overall survival time (expressed for example as posterior mean overall survival probability). U denotes the specific patient for whom we are called upon to make the choice. Z is a vector of all his covariates that can influence Y. Thus Z includes disease severity, but also other considerations such the patient’s comorbidities as well as biomarkers on the tumor cells reflecting the mechanisms targeted by the treatment. Small letters denote specific values of these variables.
While disease severity is certainly an important consideration when estimating the effect of X=x in Y, it is not enough. For example, the patient may have relatively indolent prostate cancer (disease severity) and severe cardiovascular comorbidities that could actually result in decreased survival time if we choose a therapy such as androgen deprivation therapy (ADT) as opposed to surveillance. His cancer may also harbor mutations in androgen receptor signaling (this is again different than disease severity) that can negate the effect of ADT on the cancer even though it will still yield cardiovascular toxicity.
I think these are important questions and look forward to specific clinical illustrations as answers. One thing I would add is that in your first post tagged above you plotted probabilities in the example from a model and different models (logit or log binomial for example) will return similar probabilities on the same dataset - what will differ is the value of the effect measure in strata with different baseline risks. A dataset can indeed be created that returns a constant RR or OR or RD over strata that differ by baseline risk but predictive margins when plotted from the different models on datasets with different constant effects should be illustrative.
Thank you @Pavlos_Msaouel and @S_doi. I agree that there are many ways of creating predictive models, many of which may give similar results. I also agree that there are a number of ways of measuring disease ‘severity’. However, we have to identify measures of severity that are highly predictive of the chosen outcome and which also identify subjects that will respond the treatment. Instead of using one measure of severity (e.g. the albumin excretion rate (AER) as I do) one could incorporate other measures to form a score such (e.g. the AER, HbA1c etc.) and then compare the predictive power of the calibrated score with each individual measure (e.g. AER and HbA1c). There might well be little difference, for example as the angiotensin receptor blocker (ARB) would have little effect on those diabetic patients with a high HbA1c. The baseline risk might be higher conditional on the placebo, HbA1c and the AER but the risk reduction would no greater than due to the ARB conditional on the AER alone as the ARB would not improve diabetic control.
Another central issue is how well calibrated are the probabilities of the outcome, which I don’t think featured in the recent discussion, including by @Sander_Greenland. This of course begs the question of how we should calibrate. One simple preliminary test of calibration that I use is that (1) the average of all probabilities read from a curve should match the overall frequency of the outcome and (2) the average of all probabilities read from a curve up to some point should match the overall frequency of the outcome up to that point; the same applying to those above the point. If the probabilities are not well-calibrated then we have to derive some function that does make them so. The data used to calibrate then has to be regarded as ‘training’ data’ and the calibrated curve tested on another data set. I think that the issue of calibration needs clarifying.
My journey to understand this thing called “evidence based decision making” started with a vague intuition that the norms taught to me and my colleagues were logically flawed. Since then I’ve been motivated to borrow tools from mathematical and philosophical logic to formalize this beautiful narrative description of principled and honest scientific discourse by Paul Rosenbaum in his book Observational Studies (section 1.3). Here is an excerpt, but the entire short section is worth thinking about.
I’ve learned much from Sander’s (and Frank’s) writings and posts. I credit his presentations on rebuilding statistics upon information theoretic grounds as crucial for scientific understanding.
I find it surprising that there are a large number of scholars who think practicing good science is distinct from (Bayesian) decision theoretic considerations. As a first order approximation of a rational scientific actor, an agent who attempts to maximize the information from “experiments” (defined to include observational studies) seems like a good starting point.
I acknowledge this is a minority position, but after much study, I have to disagree with the causal
inference scholars who claim probability is not a strong enough language with which to express
causal concepts. There were some interesting Twitter threads (now deleted, sadly) where Harry Crane and Nassim Taleb challenged Pearl on his position that causation is outside of standard statistical inference.
Causal inference is closely related to exchangeability, and disagreements about
study design are better discussed in terms of what factors render the groups being considered not
exchangeable.
Causal inference is just inference.[1] A community of scholars can be modeled as a group
of bettors; those who have the best models of future observations (in the sense their forecast enable
them to win more than they lose on a consistent basis). Converging to the best causal model ends
the process of betting on outcomes, unless someone finds an anomaly worth betting on, of course.
Possession of good causal models enable one to be like the gambler in JL Kelly’s paper A New Interpretation of the Information Rate.
This comment from Daniel Lakeland in that thread sums up my attitude on this elegantly:
Blockquote
I found it very frustrating to talk with Pearl regarding these issues (there was a long exchange between us on this blog about 3 or 4 years back), because I came to the conclusion just as you have that his understanding of what is probability theory and statistics is entirely frequentist … and my understanding was Bayesian… and so we talked past each other… He even acknowledged knowing about the development of the Cox/Jaynes theory of probability as extended logic, but seemed to gloss over any actual understanding of it.
The outcome in this case is a continuous variable or categorical with bins for ranges of severity. Expected values comprise this ATE: E[Y_1 - Y_0]. I understand that a physician will still want more information from the studies. Separating the ATE would seem to be useful: E[Y_1] and E[Y_0]. Nevertheless, if no other information is available, an RCT’s ATE result would seem to be useful to some degree.
One simple example of a confounder would be income. A patient with high income can afford a treatment recommendation and also is less likely to suffer from low income situations like malnutrition or financial stress. This can increase the likelihood of doing the treatment and of a positive outcome. This particular confounder may be weak and a situation may just not have any strong confounders. Then the data from the observations would not contribute much to probabilities of causation such as P(\text{benefit}) and P(\text{harm}).
Confounding in observational studies is good when estimating probabilities of causation. But a good observational study may not be feasible or available.
True, we can’t go back in time and withhold a drug that was administered (or vice-versa) to see what the two results would have been for an individual. We can only estimate ranges of probabilities of how a person will react, with and without treatment.
Yes, ideally we encapsulate these biological mechanisms in the form of a DAG and possibly functional mechanisms between variables (or at least constraints on these functions). However, in the absence of biological mechanisms, we can still compute probability ranges on probabilities of causation. Those ranges might be looser/wider without the expert knowledge, but they can often be narrow enough to make good decisions from.
Upon reading your sentence again, I think I misunderstood. With disease severity, I thought you were referring to the outcome, but now I think you were referring to a covariate. In this case, what you’d want is a Conditional Average Treatment Effect (CATE): E[Y_1 - Y_0|S], where S is disease severity. So the RCT results would either be grouped by S or a function would be fit with S as input.
Thank you @Scott for answering my questions. You are proposing different ways of reasoning with medical knowledge by invoking some principles of causal inference. @R_cubed points out that there these methods are debatable. There is also of course much disagreement amongst medical scientists when they advance alternative hypotheses based on different theories and background knowledge. The way forward of course is to conduct RCTs and observational studies to test these hypotheses to find out what happens in practice and to calibrate probabilities arrived at from RCTs with or without causal inference.
I remain concerned about the different advice that you and I would give a patient based on the result of the RCT and observational study described in your paper. You would assure a female patient that no harm can occur by choosing to take an ‘over-the counter’ drug, whereas I would warn her that it is unsafe unless taken with the same close supervision as during the RCT. I would be grateful if you could explain why we would give this different advice.
My numbers and hypothetical advice assume that the patient would administer treatment properly (at the same level as was done in the RCT). Of course, if there’s a risk of the patient not administering treatment properly then that has to be taken into account in the advice and discussion about the treatment.
But the observational study shows clear evidence that more die after choosing to take the drug than die after choosing not to take it. This implies that allowing the patient to choose ’causes’ many more to die (due to an adverse effect from one causal mechanism such as not taking the drug properly etc) that are ‘saved’ (by some other causal mechanism suggested by the RCT). Accordingly you surety cannot assume that such an adverse effect will not happen again and then conclude that a future patient choosing to take the drug has a zero probability of dying from the adverse effect. In the light of the observational study, the FDA would surely refuse a license for the proposed use (ie by allowing the patient to choose). There seems to be a divergence here between our causal inference processes! It seems that amongst other things, you may not be allowing for the possibility of more than one causal mechanism happening ‘in parallel’ at the same time.
I found a number of excellent videos on this dispute about causal inference that should be of interest.
Speaker bios
The presenters include: Larry Wasserman giving the frequentist POV, Philip Dawid, and Finnian Lattimore giving the Bayesian one.
The ones I’ve made it through so far: Larry Wasserman - Problems With Bayesian Causal Inference
His main complaint is that Bayesian credible intervals don’t necessarily have frequentist coverage. I’d simply point out that from Geisser’s perspective: in large areas of application, there is no parameter. Parameters are useful fictions. This is evident in extracting risk neutral implied densities from options prices (ie. in a bankruptcy or M&A scenario). But he discusses how causal inference questions are more challenging from a computational POV than the more simple versions seen assessment of interventions.
Philip Dawid - Causal Inference Is Just Bayesian Decision Theory
This is pretty much my intuition on the issue, but Dawid shows how causal inference problems are merely an instance of the more general Bayesian process and equivalent to Pearl’s DAGs.with the assumption of vast amounts of data.
Finnian Lattimore - Causal Inference with Bayes Rule
Excellent discussion on how to do Bayesian (Causal) inference with finite data. I’m very sympathetic to her POV that causal inference isn’t all that different from “statistical” inference, but one participant (Carlos Cinelli) kept insisting there is some distinction between “causal inference” and decision theory applications. They seem to insist that modelling the relation among probability distributions is meta to the “statistical inference” process, while that step seems naturally part of it to a Bayesian.
Panel Discussion: Does causality mean we need to go beyond Bayesian decision theory?
The key segment is the exchange between Lattimore and a Carlos Cinelli from 19:00 mark to 24:10
From 19:00 to 22:46, Carlos made the claim that “causal inference” is deducing the functional relation of variables in the model. Statistical inference for him is simply an inductive procedure for choosing the most likely distribution.from a sample. He calls the deductive process “logical” and in an effort to make Bayesian inference appear absurd, he concludes that “If logical deduction is Bayesian, everything is Bayesian inference.”
From 22:46 to 23:36 Lattimore describes the Bayesian approach as a general procedure for learning, and uses Planck’s constant as an example. In her description, you “define a model linking observations to the Planck’s constant that’s going to be some physics model” using known physical laws.
Cinelli objected to characterizing the use of physical laws to specify variable constrains when asking:
Cinelli: Do you consider the physics part, as part of statistics? Lattimore: Yes. It is modelling. Cinelli: (Thinking he is scoring a debate point) But it is a physical model, not a statistical model… Lattimore: (Laughing at the absurdity) It is a physical model with random variables. It is a statistical model.
Lattimore effectively refuted the Perlian distinction between “statistical inference” and “causal inference.” Does “statistical mechanics” cease to be part of physics because of the use of “statistical” models?
The Perlian CGM school of thought essentially cannot conceive of the use of informative Bayesian inference, which physicists RT Cox and ET Jaynes advocated.
Another talk by David Rohde with the same title of the paper mentioned above. This was from March 2022.
David Rohde - Causal Inference is Inference – A beautifully simple idea that not everyone accepts
There are still interesting questions and conjectures I got from these talks:
Why do frequentist procedures, which ignore information in this context, do so well?
How might a Bayesian use frequentist methods when the obvious Bayesian mechanism is not
computable?
How does the Approximate Bayesian Computation program (ABC) relate to frequentist methods?
This paper by Donald Rubin sparked research into avoiding the need for intensive likelihood computations that characterize ABC.
I would agree with @HuwLlewelyn and will try to summarize what @HuwLlewelyn has said thus far (please correct me if I have made a mistake Huw)
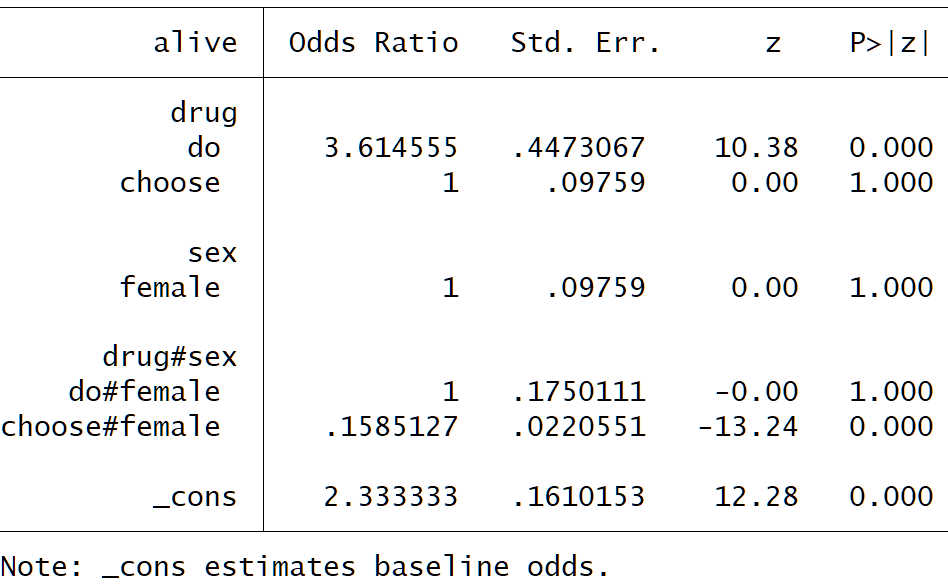
RCT
Gender has no prognostic value for the outcome
Drug decreases odds of death by 72.3%
In the trial this translates to a reduction in death from 79% to 51%
Observational study
Untreated and treated males have the same death proportion
Untreated males and untreated females have the same death proportion
Treated females have a 6.3 fold increase in odds of death
This translates to a change from 30% baseline risk of death to 73% in treated females
Interpretation
We port the RCT to the observational study thus:
a) Odds in treated males goes from 0.43 to 0.43*(1-0.723) = 0.12 then increases by choice back to 0.43 (72.3% increase). Therefore the type of male that chooses therapy is one that for some reason has equal harm to the benefit of treatment
b) Odds in treated females goes from 0.43 to (1-0.723) = 0.12 then because of choice increases to 0.43*6.3 = 2.71 i.e. an increase of about 22 fold from 0.12. Therefore the type of female that chooses treatment is one that will have a much much greater harm than the benefit of treatment
Conclusion from observational data
We need to find out what the factor related to choice is that harms both men and women but harms women much more than men
Comparison to @scott
a) First, they tell us that the drug is not as safe as the RCT would have us believe, it may cause death in a sizable fraction of patients.
I would say this does not follow from the above – rather it is the choice related factor that is the culprit here
b) Second, they tell us that a woman is totally clear of such dangers, and should have no hesitation to take the drug, unlike a man, who faces a decision; a 21% chance of being harmed by the drug is cause for concern.
This again does not follow from the data because both seem to be harmed by the ability to choose but women much more than men
c) Physicians, likewise, should be aware of the risks involved before recommending the drug to a man.
Does not seem the right decision again for a physician
d) Third, the data tell policy makers what the overall societal benefit would be if the drug is administered to women only; 28% of the drug-takers would survive who would die otherwise.
Again the data seem to suggest otherwise
I would be grateful for input from @HuwLlewelyn and @scott and happy to be corrected in these calculations but my concerns seem mostly in line with what Huw has been saying all along regarding the clinical decision making implications of these studies
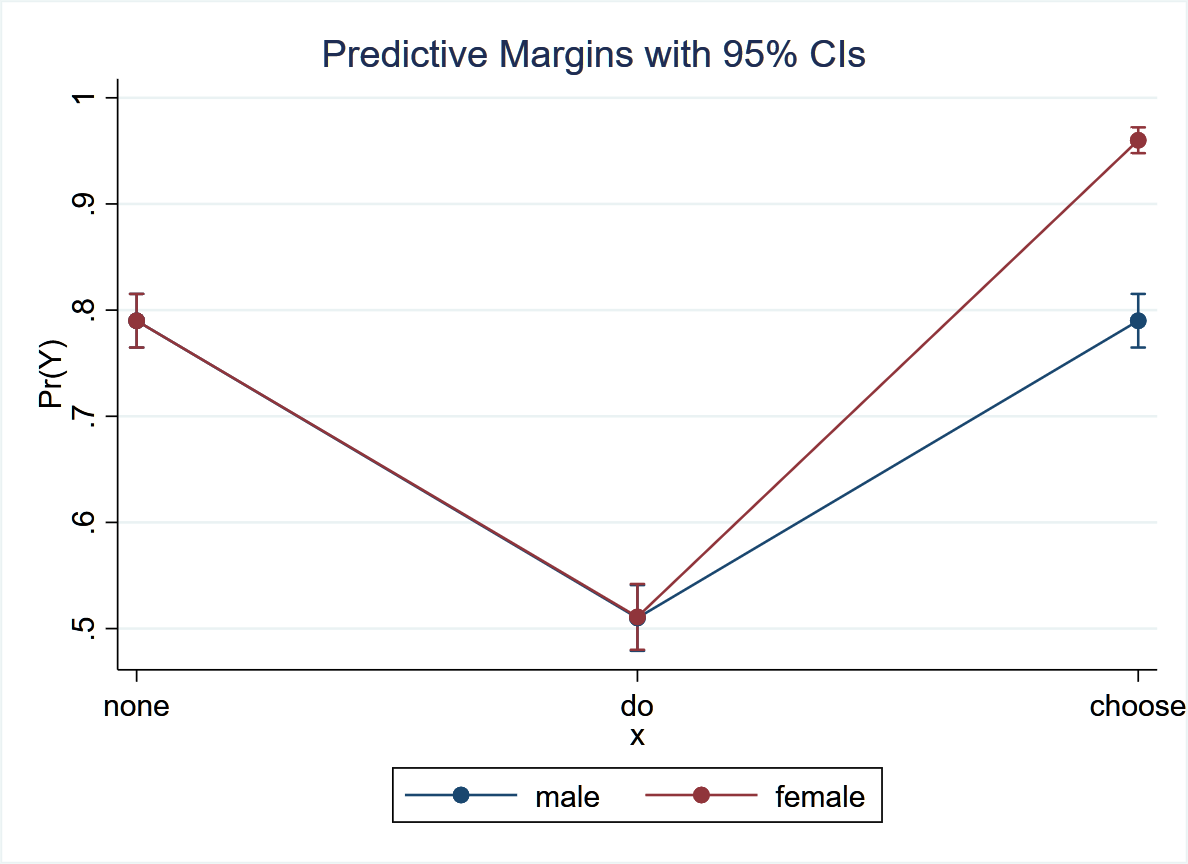
Predictive margins from logistic regression (hypothetical)
I have decided to delete the previous post and put a more intuitive one for possible comments from @scott and @HuwLlewelyn that may be more helpful in understanding the issues raised
I saw an exchange recently on Twitter and thought it was relevant to this thread. One poster wrote:
Can we express this sentence in mathematics: “similar patients given identical treatments will have different values in different studies”.
The assumption overriding counterfactuals is that Y(1, u) and Y(0,u) exist, and are immutable properties of u (the patient). Is it wrong?
Clinically speaking, the answer is “yes,” this assumption usually IS wrong when “u” is a human being. And it is exactly this fundamental misunderstanding that clinicians and statisticians find so frustrating about the hype surrounding the potential for “personalized medicine.”
Those who traffic, professionally, in stochasticity (as physicians and statisticians do), seem better placed to appreciate its scope (and therefore to give it the respect that it deserves) than those in other fields. Human biology/physiology and behaviour are each far more complex and far less predictable than a circuit board- and when human physiology and behaviour interact with each other in determining “response” to a treatment, look out- the number of possible outcomes is unfathomable.
Provided that nobody has tampered with the wiring in my house, I expect that when I turn off the breaker to my stove, it will shut off. When I flip the breaker the other way, I expect my oven to turn on. Sadly, patients are not as predictable as my oven. “Responsiveness” to a treatment is only credibly viewed as an “immutable” property of a patient in a very narrow set of clinical scenarios. Even in situations where a patient’s response to a given exposure has, historically been highly predictable (e.g., allergic reactions), responses nonetheless often attenuate over time. In oncology, where a tumour might initially respond to a treatment that blocks a biologic pathway driving the tumour’s growth, patients eventually, unfortunately, often stop responding to treatment.
Physicians have seen so many “unexpected” outcomes in their careers, that the unexpected is the only thing we have learned to expect in terms of patients’ response to treatment. If I have a patient with recurrent major depression, I am not the least bit surprised if the antidepressant that worked for her 5 years ago does not seem to work this time around. The same is true for treatment of many other conditions, including acute and chronic pain (e.g., migraine), therapies for substance abuse, epilepsy, lung disease, gynecologic disease, infectious disease…the list is endless. Rarely will a physician be surprised when a previously effective treatment does not generate the “expected” response.
An illustration of the pervasiveness of stochasticity in medicine and its impact on treatment “response:” I am not overly surprised if an otherwise healthy older patient who is anti coagulated for chronic atrial fibrillation nonetheless presents one day to the ER with a TIA. This is an “unexpected” event only in the sense that we had hoped that her anticoagulant would have made her absolute risk for TIA/stroke very low. But then, in follow-up a few days later in my office, the mystery is solved, when, on taking a careful history, the patient recalls that she had been distracted by an unplanned visit from her daughter and forgot to take her DOAC for 3 days prior to the event…
In short, human biology/physiology changes constantly and a patient’s “response” to a treatment is affected not only by these changes (which, in turn, are often affected by his environment), but also by innumerable ways in which his comorbidities interact over time, by his behaviour/decisions (in which case, physiologic complexity is effectively multiplied by behavioural complexity), and by innumerable stochastic factors that are part of everyday life. Physicians know that there are very few “immutable properties of u.”
I agree that this Tweet was imperfectly phrased, and that it makes an unconvincing and strong claim about deterministic counterfactuals. I had anticipated that there would be responses such as this one, which are very reasonable, and I had already planned to add my thoughts on Twitter. I can no longer find the original tweet, it appears as if the author may have recognised that it could be misinterpreted and deleted it (?). Either way, thank you for giving me an opportunity to respond here instead, without being constrained to 140 characters!
When using causal models, you can either rely on an ontology with deterministic counterfactuals (as in the tweet) or one with stochastic counterfactuals (where every individual u has an outcome that is drawn randomly from their individual counterfactual distributions f_u(Y_u)(1) and f_u(Y_u)(0)). My understanding is that almost all the foundational results in causal inference are invariant to whether the causal model uses deterministic or stochastic counterfactuals, but that it is sometimes didactically useful to focus on deterministic variables, which are often easier to understand. Given that the results hold under either model, this simplification often appears justifiable, not least because it significantly reduces notational load.
I fully agree that treatment outcomes are highly situational and often unpredictable, that they depend on a large number of factors which are too complex to attempt to model explicitly and therefore best understood as “stochasticity”. But the crucial point I want to make, is that if we want to make decisions to optimize outcomes, it is necessary to find a structure to the randomness: If everything was fully random (without any structure), we could never have any rational reasons for preferring one treatment option over another.
Stochastic counterfactual distributions are ideally suited for representing the relevant kind of structure. If a person’s outcomes depend on a specific aspect of physiological complexity, there are some settings where this may be irrelevant to your decision making (such that it is acceptable to consider it “randomness” or “noise”), and other settings where causal reasoning will reveal that it needs to be tackled heads on in the analysis. Only with a causal model is possible to fully clarify the reasoning that optimises predicted outcome under intervention, and these causal models will use something like a counterfactual distribution to represent the structure that it imposes on the stochasticity.
I don’t think anyone in causal inference expects treatment response to be constant over time. When they talk about counterfactuals as “immutable personal characteristics”, I believe they are talking about a highly situational construct that is in some sense “known to God” and that represents what will happen to the patient if they take treatment at some specified time, but which is certainly not assumed to be stable over time. At the very least, the original Tweet should have given a time index to the counterfactuals.
The distinction between stochastic and deterministic counterfactuals is philosophically very interesting, and there may exist multiple rational ways to think about these constructs. I think it was a mistake for the original tweet to imply that causal inference depends on a “realism” about deterministic counterfactuals.
Wonderful post. @Stephen has written extensively about this. My simple take on this work is this:
If you enter the same patient multiple times in a clinical trial and each time measure their change from baseline in systolic blood pressure you’ll get a different change each time. Patients are inconsistent with themselves so there is a limit to “personalization”.
If you did a 6-period 2-treatment randomized crossover study you can estimate the treatment effect for individual patients if blood pressure is measured precisely enough. That means that we can estimate the long-term tendencies for a patient (average of 3 measurements per treatment). But we still can’t estimate very well a patient’s current singly-measured blood pressure.
I think this is really the crux of the issue. Some might argue that trying to find structure in randomness is to deny the very existence of randomness…Opinions about whether or not it’s futile to even try to find patterns in chaos seem (crudely) to delineate two ideologies- causal inference epidemiology and statistics.
In fairness, I don’t think that views of the two camps are as black and white as this. Plainly, causal inference proponents don’t deny the existence of randomness and statisticians don’t deny the possible existence of occult cause/effect relationships in the world. Rather, the two groups seem to differ in their opinions about what type(s) of evidence will allow us to make the best “bet” when we are treating our patients, with a view to optimizing their outcomes.
Causal inference epidemiologists seem to feel that the world isn’t quite as random as many believe, and that if we can chisel out even a few more cause-effect relationships from the apparent chaos, then maybe we can make better treatment decisions (?) Maybe there’s some truth to this view, but if the number of clinical scenarios in which it might apply is small (e.g., conditions with strongly genetically-determined treatment responses), then the cost/effort involved in trying to identify such relationships could easily become prohibitive. In the long run, addressing social determinants of health would likely pay off much more handsomely with regard to improving the health of societies. Seeing governments/research funders throw huge sums of money toward what many consider to be a fundamentally doomed enterprise is aggravating, to say the least.
But getting back to the paper linked at the beginning of this thread: Maybe I’m misconstruing, but the authors seem to believe that it’s possible to infer treatment-related causality for individual subjects enrolled in an RCT, simply by virtue of the fact that they had been randomly assigned to the treatment they ended up receiving. For example, they seem to assume that anybody who died while enrolled in a trial must have died as a direct result of the treatment (or lack thereof) he received during the trial. In turn, it seems that this belief stems from a more deep-seated conviction that a patient’s probability of “responding” to a treatment is somehow “predestined” or engrained in his DNA, and therefore will be consistent from one exposure to the next. People who have posted in this thread are trying to point out that this conviction is incorrect. And if this is the underlying assumption on which the promise of “personalized medicine” hinges, then health systems are throwing away a whole lot of money in trying to advance the cause.
If you were to review case reports for all subjects who died (unfortunately) while they happened to be enrolled in a very large, longterm clinical trial, you wouldn’t be surprised to find some subjects who died from airplane crashes, slipping on banana peels, pianos falling from the sky, aggravated assault, accidental drug overdose, electrocution, and myriad medical conditions that were completely unrelated to the treatment they received (e.g., anaphylactic reaction to peanuts, bacterial meningitis outbreak, forgetting to take an important medication…). Events like this are recorded in both arms of clinical trials and have nothing to do with the treatment being tested. Presumably, though, the more deaths that are recorded, the more convinced we can be that between-arm differences in the proportion of patients who died might be due, at a group level, to the treatment in question, rather than simple bad luck.
Even if the tweet referenced above is now deleted, there’s plenty of circumstantial evidence that the author fundamentally believes that patients can be viewed like circuit boards- as though they are intrinsically “programmed” (like a computer) to respond the same way, whenever the “input” is the same.
I’m not a statistician nor an epidemiologist. So I don’t know how to phrase what I’m trying to say using math. But after practising medicine for 25 years, I’m not sure that it’s possible, for physicians to make a better “bet” (in most, but not all, cases) regarding the treatments we select for patients than one that is grounded in the results from well-designed RCTs. This approach seems completely rational to me. Conversely, I perceive innumerable ways that we could fool ourselves in the process of trying to identify engrained “individual responses” in a sea of potentially random events.
This needs clarification: There are at least two major types of probabilities used in stochastic inference:
Aleatory probabilities that are connected to physical processes such as the random treatment allocation in randomized controlled trials (RCTs). This is randomness that is based on a well-defined physical process and its uncertainty can thus be validly quantified by standard statistical methodology.
Epistemic probabilities that express our ignorance.
The two can be numerically equivalent and considered to express “randomness”. But they are fundamentally different as nicely described, e.g., here.
Because frequentism focuses on aleatory probabilities whereas Bayes allows both, these considerations can degenerate into the endless frequentist vs Bayes debate that would be counterproductive in this thread. Whether using a Bayesian or frequentist lens, a major task for physician scientists is to convert epistemic probabilities into aleatory ones as much as possible, chiefly through experimental design along with careful observations such as correlative analyses of patient samples. These need to be embedded in statistical models informed by causal considerations.
Almost every procedure used by “classical” statisticians imposes structure on randomness. Without such structure, there would be no way to do any kind of inference.
The difference between classical statisticians and causal inference epidemiologists, is not that epidemiologists assume “more” structure and “less” randomness. In practice, we use the same estimation procedures, and to the extent that we have different preferences about model choice, those differences do not reflect different allowances for “randomness”.
Rather, the difference is that we (that is, epidemiologists) insist on having a language for reasoning about whether the assumptions we make about the structure of randomness are consistent with our beliefs about how reality works, so that those assumptions can be evaluated as an integral (and essential) part of the overall scientific inferential procedure.
Without cause-effect relationships, statistics gives absolutely no basis for making rational decisions, you might as well read tea leaves. Cause and effect is there whether we believe in it or not. We can either tackle this heads on with a scientific language for determining whether and how we can learn about causal effects from the data, or we hide our heads in the ground and hope that we magically get the right causal answer from non-causal statistical inference.
There are some statisticians who deny the usefulness of the counterfactual language. In my view, they are invoking the magical category of “randomness” to sweep the issue under the carpet, unilaterally declaring that their preferred modelling approach is the canonical one-size-fits-all procedure for imposing structure on randomness, even when alternatives are just as consistent with living in a stochastic world.
Finally, I want to note that despite its exaggerated claims of importance, the paper that that is discussed in this thread is a very idiosyncratic approach that can only be used in some highly artificial settings, and even then, with highly questionable utility. It is most certainly not an accurate summary of current thinking in personalized medicine, and it does not reflect consensus among the causal inference crowd.
This depends on what you mean. When applied to a causal design (e.g., a randomized experiment where there is no post-randomization trickery) causal language is hardly needed at all.
The real problem with causal epidemiology is when the rubber hits the road. Lots of methodologists talk about notation and theory but can’t give us a real complete case study based on real data – a case study in which the DAG is justified by the subject matter and all needed measurements are available in the data. A case study where the rest of us can learn how to do real and not theoretical causal inference. See the call for examples here.